デュシェンヌがた‐きんジストロフィー【デュシェンヌ型筋ジストロフィー】
読み方:でゅしぇんぬがたきんじすとろふぃー
X染色体上にあるジストロフィン遺伝子の異常により発症する進行性筋ジストロフィーの一つ。筋細胞の構造を保つ役割を担うジストロフィンが生成されず、筋肉が崩壊と再生を繰り返しながら萎縮していく。出生男児約3500人に一人の割合で発症する。3〜5歳頃に転びやすい、走れないなどの症状が現れ、10歳前後で歩行不能となることが多く、進行とともに呼吸機能の低下や心不全などの重篤な症状を示す。病名はフランスの神経学者デュシェンヌに由来。DMD(Duchenne muscular dystrophy)。→ベッカー型筋ジストロフィー
[補説] 筋細胞の崩壊に伴ってクレアチンキナーゼ(CK)の血中濃度が上昇するため、発症前の乳児期に血液検査で発見されることもある。
デュシェンヌ型筋ジストロフィー
英訳・(英)同義/類義語:DMD , Duchenne muscular dystrophy, Duchenne muscular dystrophy
ジストロフィンタンパク質を指令するX染色体上のDMD遺伝変異による劣性疾患で、骨格筋と心筋の変成を起こす。ベッカー型筋ジストロフィーは同じ遺伝子の変異による軽症型。
デュシェンヌ型筋ジストロフィー
本症はX連鎖(性染色体)劣性遺伝をとるため、患者は男児に限られます。同じ様な遺伝形式をとる病気に血友病や色盲があります。母親が遺伝子に異常(変異)をもっていて、それが男の子に伝わることが多いのです。しかし、母親が遺伝子変異をもっているとは限りません。この病気は突然変異率が高いので、お子さんの遺伝子に突然変異があり、お母さんには変異がないことがまれではありません。お母さんの約1/3は変異がありません。
まれに染色体異常(X染色体と常染色体の相互転座、Turner症候群)があると、女性も同様な症状をとります。また女性保因者(女性で遺伝子の異常を持っている人)も、まれですが血液のCK値が高かったり、軽い筋肉の力が弱かったりします。
この病気を起こす遺伝子はどこにあって、その遺伝子に変異があるとどのような蛋白が欠損するか明らかになっています。遺伝子のある場所はX染色体短腕(Xp21)にあります。この遺伝子は分子量427kDの蛋白(ジストロフィンと命名されています)をコードしています。ジストロフィン遺伝子はクローニングされ、それはcDNAで14kbあって、79のエクソンからなっています。患者さんではこのジストロフィン遺伝子に変異があり、ジストロフィン蛋白が生成されないのです。患者さんの50ー60%はジストロフィkン遺伝子の欠失(遺伝子が欠けていて短い)、約10%は重複(遺伝子が重なって長くなっている)です。残りの30ー40%は多分点変異(DNAの一個が間違っているような一塩基置換など)と考えられています。 最も多い欠失は、3塩基づつの読みにずれがある(out of frame)欠失(正常では塩基3個で一つのアミノ酸ができます。3個の組み合わせがずれるので、めちゃくちゃなアミノ酸ができるのです)です。それだけでなく、欠失部以下にもう蛋白を作るのを止めなさいというストップ命令(コドン)が働き、不完全な蛋白が生成されます。それは不安定で、すぐに分解されると考えられています。ですからデュシェンヌ型ではジストロフィン蛋白はまったく作られません。
ジストロフィン蛋白は筋細胞膜直下に局在していて、ジストロフィン結合蛋白と結合しています。ジストロフィンは細長い棒状構造をしていると考えられています(図6)。
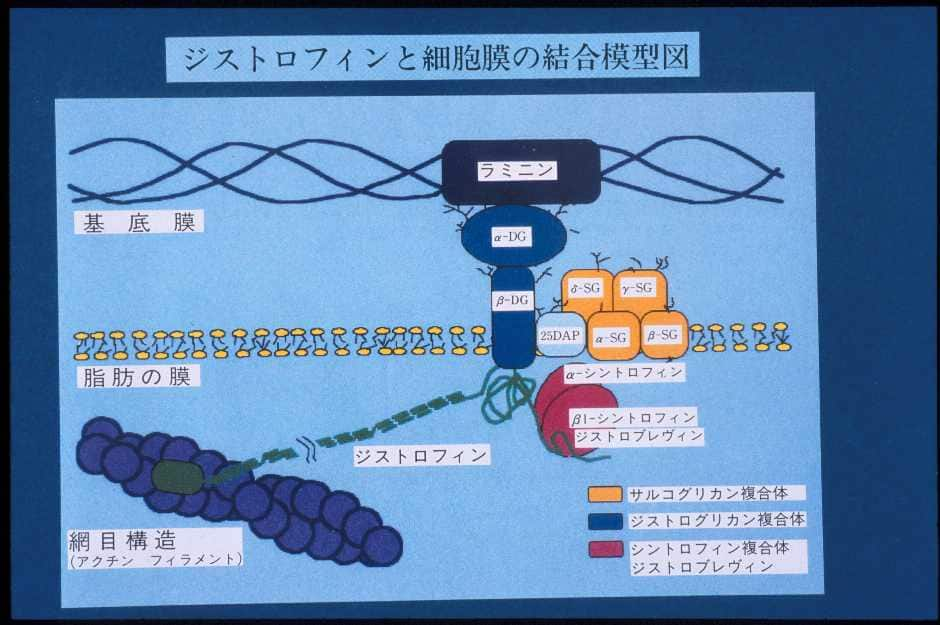 |
| 図6:ジストロフィンとジストロフィン結合蛋白の関係 この図は筋細胞膜(基底膜と形質膜の2重膜からなる)の一部を拡大した分子モデルの図である。 20DAPはサルコスパンとよばれている。DG:ジストログリカン、SG:サルコグリカン |
5’側(ジストロフィンの頭の部分)は細胞膜をしっかりと強固に保つ、アクチンという細い糸と結合する部位(actin binding domain)で、それに続いて3重らせん構造をとる桿状部分(rod domain)が続いています。それはさらにシスティンに富む部分(cysteine-rich domain)、C端(carboxyl terminal)へと続いています。図6のようにジストロフィン分子はcysteine-rich domainのところでジストロフィン結合蛋白のβジストログリカン(β-dystroglycan)と結合しています。ジストロフィンはアクチンという細い線維と一緒になって膜を補強するものです(細胞膜をしっかりと裏打ち構造をするもので細胞骨格といいます)。
患者さんではこのジストロフィンは全く欠損しています。それは抗ジストロフィン抗体で免疫染色をすると、正常では筋細胞膜に局在するジストロフィンが全く染色されないことから容易に判定でき(図7)、本症の診断用に使用されています。細胞膜をしっかりと安定させているジストロフィン蛋白がないので、細胞膜は弱く、すぐにシャボン玉のようにこわれるのでしょう。
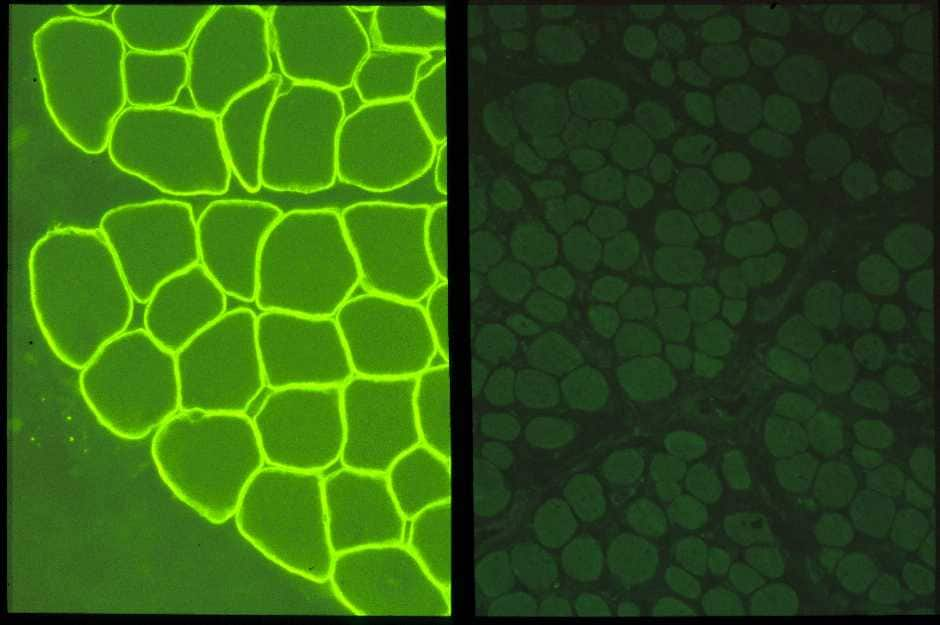 | 正常筋(左)では筋細胞膜にジストロフィンが存在するので、膜の部分が蛍光を発し、染色されている。 デュシェンヌ型(右)ではジストロフィンがないので、まったく染色されていない。 |
| 図7:ジストロフィン抗体による免疫染色 | |
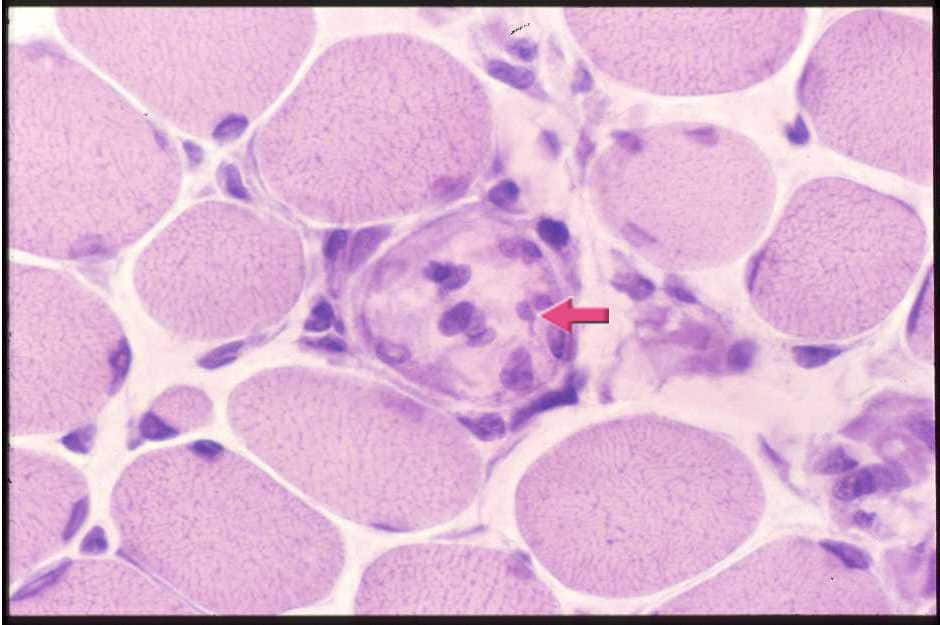 | 筋線維の大小不同と壊死線維(この図の中央にあって、溶けたような胞体をもっている)(矢印)をみる。 壊死線維には大型の核をもつマクロファージが侵入している。 |
| 図8:デュシェンヌ型筋ジストロフィーの筋病理-1 | |
| 筋ジストロフィーにみられる沢山の再生線維。中央にみえる青みを帯びた小さな線維(→) で、デュシェンヌ型では筋線維の平均15%は再生途上筋である。 マクロファージで掃除され、きれいになった壊死細胞には続いて再生が起こる。病気が進むと筋線維は著明に減少し、結合織と脂肪織で置換されてしまう。こうなると力は弱く、関節が硬くなり、伸びなくなってしまう。 | 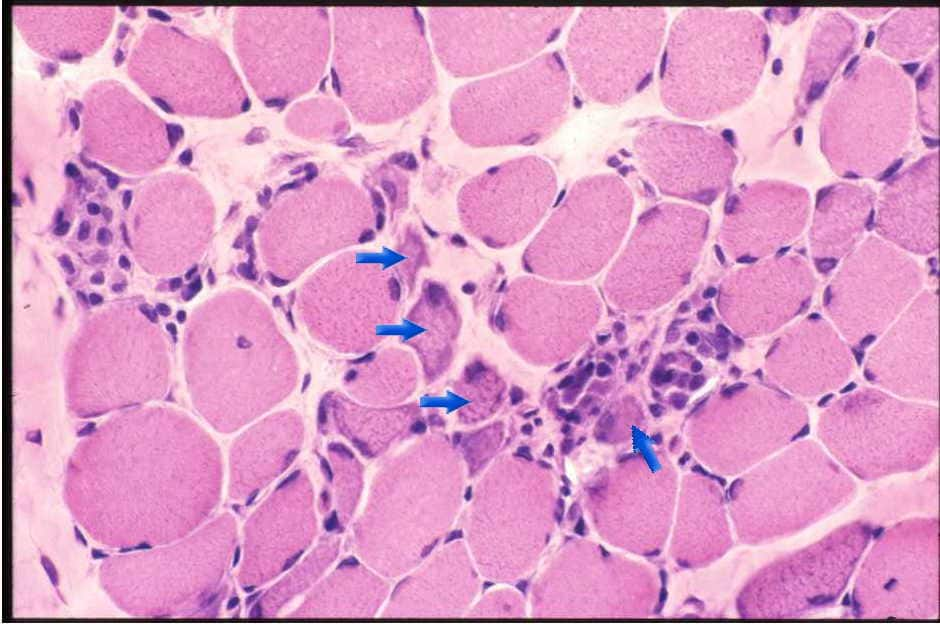 |
| 図9:デュシェンヌ型筋ジストロフィーの筋病理-2 | |
b.臨床症状
本症の頻度は男児出生3、300人に1人、人口10万人に3ー5人くらいの患者さんがいるといわれています。人種差はなく、あらあゆる国に患者さんがいます。
従来は3ー5歳頃、走れない、転びやすい、階段の昇降困難で気付かれることが多かったのです。しかし最近では、乳児期に他の疾患(たとえば風邪をひいたとか)での検査中に偶然高CK血症で見いだされることが多くなりましたた。乳児期から追跡すると歩行開始遅延(1歳6カ月以降に歩行開始)が30ー50%いること、歩行開始時にはすでに筋力低下(立ち上がり方の異常)がみられることから、乳児期にもすでに異常があるお子さんがいるようです。
病初期は上記のように転びやすい、走れないなど歩行に関する異常が最も多くみられます。また筋肉の力が明らかに弱くなると、そん居の姿勢から立ち上がるとき、床に手をつき、臀部を高く挙げて立つようになります。健康なお子さんは、しゃがんだ位置からすっと垂直に立ち上がります。さらに筋肉の力が弱くなると、手を床に着き、つぎに膝に手を交互にあてて立つ、いわゆる登はん性起立(Gowers徴候)をみるようになります
(図10)。
| 腰の筋力低下があるため、床から起立する時、まず床に手をついて、お尻を高くあげる(a)。 次にひざに手をあてて、手の力を借りて立ち上がる(b)。 ふくらはぎは太く偽性肥大を示している。 この偽性肥大はデュシェンヌ型、ベッカー型に特徴的である。 | 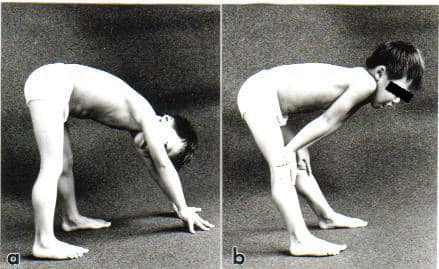 |
| 図10: 登はん性起立(Gowers徴候) | |
さらに進行すると、何かものにつかまらないと立てなくなり、10歳前後で歩行不能となり、車椅子生活となります。次第に寝返りにも人の介助が必要となり、20歳前後で、呼吸筋の力が弱くなるため、人工呼吸器の助けが必要となります。むかしは人工呼吸器がなかったので、患者さんは20歳以前に死亡していました。でも今は40歳まで生きる方がでてきました。医療機器の進歩で、患者さんの生命はもっともっと伸びるでしょう。
筋萎縮(見た目に筋肉が細くなる)は病初期にはあまり目立たちません。むしろふくらはぎが異常に太いのが特徴的で、これは仮(偽)性肥大(pseudohypertrophy)とよばれています。この筋の肥大は肩や、頬筋、舌筋にもみられます。ふくらはぎの肥大はデュシェンヌ型や次に述べるベッカー型では、ほとんどの患者さんが経験する病気に特徴的所見です。
病気が進行すると、筋萎縮は躯幹近位筋(大腿、上腕、躯幹筋)に著明にみられるようになります。歩行時には関節拘縮(関節の伸展が悪くなることです)はアキレス腱の短縮による尖足のみですが、歩行不能となった時点から、股関節、膝関節などに広がっていきます。脊柱変形、手指、顎関節など全身の関節の拘縮をみるようになるのです。腱反射はアキレス腱反射を除いて減弱ないし消失します(図11)。
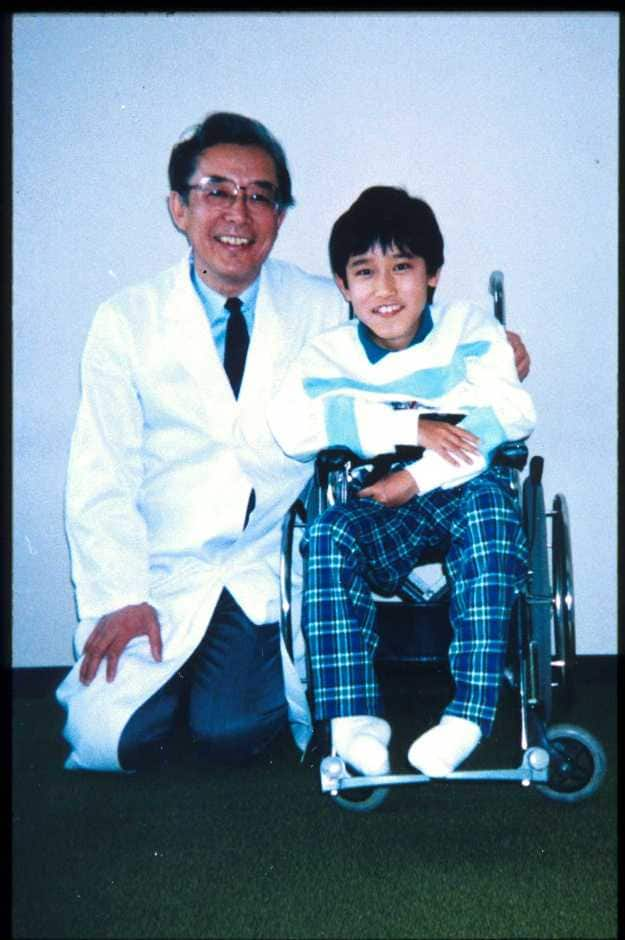 | 足の関節が拘縮している。 T君は今人工呼吸器をつけているが、亜細亜大学法学部に通学し、勉強しているファイトあふれる青年(この写真の掲載はT君の許可を得たものです)。 |
| 図11:車イス生活になったデュシェンヌ型筋ジストロフィーのT君と筆者 | |
現在のところ病気の進行をとめたり、筋力が回復するような根本的治療法はみつかっていません。でも、病気の進行を遅らせるさまざまな試みがなされています。
その一つとして副腎皮質ホルモン投与が試みられています。副作用が出ない少量投与でよい結果が出ています。 わたしはデュボビッツ(Dubowitz)先生の方法(毎月10日間だけプレドニゾロン0.75mg/Kg/日投与、20日間休薬)をとっています。 これですと副作用が出る人はごくまれです。副作用もほっぺがふっくらとする程度です。 中にはとても効果があって、5年くらい病気の進行がなくなる人もいます。 でも、効果がはっきりしない人もいます。アメリカの調査では副腎皮質ホルモン治療を受けた人は歩行期間が2年も延長するとのことです。
最近、アメリカの神経学会、小児神経学会では多くの患者で治験をしたところ、最初はプレドニソン0.75mg/Kg/日の連日投与から開始することがもっとも有効であったとして、推薦しています。副作用が出てきたら、0.3mg/Kg/日に減量します。(http://www.aan.com/professionals/practice/pdfs/DMD_Guideline_Physicians.pdf)。 この量ですと、日本人には量が多すぎて、副作用が出る可能性が強いと思います。日本人用のスタンダードが欲しいです。
デュシェンヌ型では筋力低下、関節拘縮が進行するので、それを少しでも防ぐためのリハビリテーションが行われ、効果をあげています。 また呼吸不全をみるようになった場合は鼻マスクによる人工呼吸器(nasal:noninvasive intermittent positive pressure: NIPP)の使用、あるいは気管切開による人工呼吸器の使用が行われ、患者さんの延命効果に大きく寄与しています。 心不全徴候がみられるようになった患者さんには、アンジオテンシン変換酵素阻害剤(ACE阻害剤)、ベーターブロッカー、ジギタリス剤、利尿剤を組み合わせた治療が行われていて、とてもよい結果をえています。 いろいろな薬物治療、呼吸管理などで、患者さんの生命予後は飛躍的によくなっています。
遺伝子治療はmdxマウス(ジストロフィンが欠損している筋ジストロフィーマウス)を使用して行われています。 現在アデノウィルスDNAをベクター(DNAの細胞内への運びやさん)にして、ジストロフィン遺伝子の一部あるいは全長が挿入され、筋内投与が行われています。 投与部にはジストロフィン陽性線維が増加し、筋線維の壊死の抑制がみられています。ただし、1回投与での効果の持続性が短く、再投与での効果に乏しいことが欠点とされています。 でもアデノウィルスベクターに変わるベクター(たとえばアデノウィルス随伴ウィルスベクター)なども開発されています。 遺伝子治療はよいベクターの開発にかかっています。科学の進歩はすばらしいです。原因がまったく分からなかったデュシェンヌ型筋ジストロフィーの遺伝子がクローニングされたのが1986年です。 最近の学問の進歩には目を見張るものがあります。根本治療、遺伝子治療への時代に突入したのです。
遺伝子治療については別に、国立精神・神経センター神経研究所 遺伝子治療研究部 武田伸一部長に解説いただいています。
→デュシェンヌ型筋ジストロフィーに対する分子治療のこころみ
デュシェンヌ型筋ジストロフィー
出典: フリー百科事典『ウィキペディア(Wikipedia)』 (2024/11/06 01:41 UTC 版)
| Duchenne muscular dystrophy | |
|---|---|
 |
|
| デュシェンヌ型筋ジストロフィーの患者のふくらはぎの筋肉の断面の顕微鏡像。筋線維が広範囲にわたって脂肪細胞に置き換わっている。 | |
| 概要 | |
| 診療科 | 小児神経学, 神経筋医学, 遺伝医学 |
| 症状 | 筋力低下、起立困難、脊椎側彎症[1][2] |
| 発症時期 | 4歳前後[1] |
| 原因 | 遺伝(性染色体劣性)[2] |
| 診断法 | 遺伝子検査[2] |
| 使用する医薬品 | 副腎皮質ホルモン |
| 治療 | 薬物治療、理学療法、装具療法、言語聴覚療法、作業療法、外科手術、補助換気[1][2] |
| 予後 | 平均寿命: 26歳[3] |
| 頻度 | 3,500-6,000人に1人(男性)[2] 50,000,000人に1人(女性)[4] |
| 分類および外部参照情報 | |
| Patient UK | Duchenne muscular dystrophy |
デュシェンヌ型筋ジストロフィー(デュシェンヌがたきんジストロフィー、英: Duchenne muscular dystrophy、略称: DMD)は、主に男児が影響を受ける重症型の筋ジストロフィーである[2]。通常、筋力低下は4歳前後から始まり、急速に悪化する[1]。筋肉の喪失はまず大腿や骨盤周囲の筋肉、続いて腕でみられるようになり、その結果起立困難となる[2]。患者の大部分は12歳までに歩行が困難になる[1]。疾患の影響を受けた筋肉は、脂肪量の増加のために大きくなったように見える場合がある[2]。脊椎側彎症もまた一般的である[2]。患者の一部では知的障害も見られる可能性がある[2]。欠陥のある遺伝子を1コピーだけ持つ女性は保因者となるが、軽度の症状がみられる場合もある[2]。
この疾患は性染色体劣性遺伝する[2]。疾患の原因はジストロフィンをコードする遺伝子(DMD遺伝子)の変異であり、症例の約2/3は母親からの遺伝、残りの1/3は新たに獲得された変異が原因である[2]。ジストロフィンは筋線維の細胞膜の維持に重要な役割を果たすタンパク質である[2]。この疾患は遺伝子検査によって出生時に診断が行われることが多い[2]。また、患者の血液では高レベルのクレアチンキナーゼがみられる[2]。
この疾患の根本的な治療法は知られていないが、一部の症状には理学療法、装具の使用、矯正手術が有用である場合がある[1]。呼吸筋の筋力が低下した患者では、補助換気が必要となる場合がある[2]。投薬としては、筋変性を遅らせるためのステロイド、痙攣や一部の筋肉の活動を制御するための抗痙攣薬、死滅しかかっている筋細胞への損傷を遅らせるための免疫抑制剤などが投与される[1]。遺伝子治療に関してはヒトで初期段階の研究が行われており[2]、小規模な初期研究では一部の小児で筋力の改善がみられているが、2020年の時点で長期的な影響は不明である[5]。
DMDは3,500人から6,000人の男児に1人の割合で発生する、最も一般的なタイプの筋ジストロフィーである[2]。平均寿命は26歳であるが[3]、良好なケアを行うことで30代や40代まで生存する場合がある[2]。この疾患は女児では極めて稀であり、約5000万人に1人の発生率である[4]。
徴候と症状

DMDでは、筋線維の錯綜、細胞死、結合組織や脂肪への置換を原因とする進行性の筋力低下が引き起こされる[2]。まず随意筋、特に臀部、骨盤周囲、大腿、下腿の筋肉に影響が生じる[6][7]。次第に肩や首、その後に腕、呼吸筋やその他の部位に進行する[7]。疲労も一般的な症状である[8]。
通常、疾患の徴候は5歳以前に出現し始め、歩き始めた時点からすでに観察される場合もある[9]。運動技能は一般的に困難であり、ぎこちない歩き方や走り方となる[10]。また、つま先歩きになる傾向があり[10]、その一因はアキレス腱の短縮[11]や膝伸筋の筋力不足である[7]。転倒も頻繁に起こる[12]。成長に伴って歩行はますます困難なものとなり、通常13歳までに歩行は完全に不可能になる[10]。DMDの患者の男性の大部分は、21歳までに基本的に首から下が麻痺した状態となる[9]。心筋症、特に拡張型心筋症も一般的であり、18歳の患者の約半数にみられる[10]。うっ血性心不全や不整脈の発生は時にみられる程度である[7]。疾患の後期段階では、呼吸障害や嚥下障害が生じ、肺炎の原因となる場合がある[13]。

DMDの典型的な徴候は臥位または座位からの起立困難であり[12]、ガワーズ徴候と呼ばれる特有の立ち上がり方が観察される。この立ち上がり方は子供がうつぶせの状態から起き上がろうとする際に腕を用いて骨盤周囲の筋力不足を補う方法であり[10]、まず腕と膝で立ち、脚をよじ登るうように手を動かすことで直立する。他の特徴的な徴候には、舌、下腿、臀部、肩の筋肉の仮性肥大がある(4–5歳でみられる)。筋肉組織が最終的に脂肪や結合組織に置換される現象であるため、「仮性」肥大と呼ばれる。アキレス腱やハムストリングで筋線維の変形や拘縮が生じ、筋線維の短縮や結合組織の線維化のために機能が障害される[7]。腰椎過前彎、脊椎側彎、骨盤前傾、胸郭変形など、骨格の変形も生じる場合がある。腰椎過前彎は、臀筋や大腿四頭筋の筋力低下に対する代償機構と考えられており、こうした変化はすべて姿勢や歩行の変化(股関節の伸展の制限など)を引き起こす[14][15]。
DMDでは筋肉や骨格以外にも症状が見られる。神経行動障害(ADHDなど)、学習障害(ディスレクシアなど)、特定の認知機能(特に短期言語記憶)の非進行性の低下のリスクが高い[10]。これらは脳内のジストロフィンの欠乏もしくは機能不全によるものであると考えられている[16]。
DMDの発症時には、血液脳関門の破壊も特筆すべき特徴となる[17]。
DMDの患者では血漿中のリポタンパク質濃度の上昇がみられ、原発性脂質異常症であることが示唆されている[18]。
原因

DMDは、X染色体の短腕(Xp21)に位置するジストロフィン遺伝子の変異によって引き起こされる[19]。変異によって、筋細胞の構造的完全性を維持しているジストロフィンタンパク質の量が大幅に減少したり、存在しなくなったりする[20]。ジストロフィンは、各筋線維のアクチン細胞骨格を周囲の基底板(細胞外マトリックス)へ、多くのサブユニットからなるタンパク質複合体を介して連結する。ジストロフィンが存在しない場合、過剰量のカルシウムが筋鞘(細胞膜)を透過する[21]。ミトコンドリアの機能不全によって細胞質基質でのストレス誘導性のカルシウムシグナルが増幅され、活性酸素種の産生が増幅される[21]。いくつかの経路が関与するこの複雑なカスケードは明確には理解されていないが、細胞内の酸化ストレスの増加によって筋鞘が損傷し、最終的には細胞死が引き起こされる。筋線維は壊死し、脂肪組織や結合組織に置き換えられる。

DMDはX連鎖劣性遺伝する疾患である。患者の母親は2本のX染色体のうち1つが欠陥遺伝子であり、欠陥のあるX染色体を受け渡すことで息子に疾患が引き起こされる。男性は母親からX染色体を、父親からY染色体を受け継ぐため、保因者である母親から生まれた息子は50%の確率で疾患を発症する。保因者の娘も50%の確率で欠陥遺伝子を受け継ぐが、女性は両親からX染色体を1本ずつ受け継ぐため、父親から受け継いだ正常なX染色体が母親から受け継いだ欠陥を補償し、影響が生じることは通常はない。患者は父親としてX連鎖疾患を息子へ受け渡すことはないが、娘はその疾患の保因者となる。また、保因者の女性でも軽度の症状がみられることがある[2]。
こうした理由により、DMDは女性では極めて稀である(約5000万出生につき1人)[4]。父親が患者で母親が保因者である場合や、X染色体を1本欠損している場合、X染色体の不活性化(最も一般的)などによって、女性にも出現する場合がある[22]。父親が患者で母親が保因者である場合、父親からは常に欠陥X染色体を受け継ぎ、母親からは50%の確率で欠陥X染色体を受け継ぐため、等確率で患者または保因者かとなる[23]。
診断
DMDの疾患の家族歴がある人には遺伝カウンセリングが勧められる。DMDは妊娠時の遺伝子検査によって約95%の確度で検出することができる[13]。
DNA検査
ジストロフィン遺伝子の筋特異的アイソフォームは79個のエクソンから構成され、DNA検査(血液検査)と解析では、影響を受けているエクソンの変異の種類が同定される。大部分の症例で、DNA検査によって確定診断となる[24]。
筋生検
DNA検査で変異を見つけることができなかった場合には、筋生検が行われる可能性がある[25]。生検針を用いて、少量の筋組織が採取される。DMDの生検試料に対して行われる重要な検査は、ジストロフィンに対する免疫組織化学、免疫細胞化学、イムノブロッティングによる検査であり、経験豊富な神経筋病理医によって解釈がなされるべきである[26]。これらの検査からは、ジストロフィンタンパク質が存在するかに関する情報が得られる。タンパク質が存在しないことはDMDを意味し、存在する場合には検査によってその存在量や分子量が示されるため、DMDとより軽症のジストロフィノパチーとの鑑別に役立つ[27]。過去数年の間に、DNA検査は疾患の原因となる変異をより多く検出できるよう進歩しており、DMDの確定のための筋生検はかつてほどは必要とされなくなっている[28]。
出生前診断
母親が保因者であるまたは保因者であることが疑われる場合には、出生前診断が考慮される[29]。
男児はDMDである可能性があるが、女児のDMDは極めて稀であるため、侵襲的な検査を行う前に胎児の性別を明らかにすることが重要である。性別の判定は16週で超音波検査によって行うことができ、また近年では胎児由来セルフリーDNA検査によっても行うことができる。絨毛採取(CVS)は11–14週の間に行うことができ、流産のリスクが1%ある。羊水検査は15週以降に行うことができ、流産のリスクが0.5%ある。新型出生前診断(NIPT)は11–14週前後に行うことができる[30]。
治療
DMDの根本的治療法は存在せず、医学的需要は規制当局に認識されている[31]。遺伝子治療の臨床試験は一部成功を収めている[32]。
治療は一般的には症状の制御を目的としており、質問票を用いて測定されるQOLを最大化することが目的となる[33]。次のような治療が行われる。
- プレドニゾロンやデフラザコートなどの副腎皮質ホルモン剤は、最大2年間にわたって筋強度の短期的改善をもたらす。副腎皮質ホルモン剤は歩行可能期間を延ばすことも報告されているが、そのエビデンスは強固ではない[34]。
- 理学療法は筋肉の強度、柔軟性、機能の維持に有用である[2]。
- 疾患の進行に伴い、適切な呼吸補助を行うことが重要である[2]。
- 心臓の問題により、ペースメーカーが必要とある可能性がある[35]。
モルフォリノアンチセンスオリゴ医薬品エテプリルセンは、アメリカ合衆国でジストロフィン遺伝子のエクソン51スキップに応答する患者に対して承認されている。エテプリルセンは臨床的ベネフィットが確立されなかったため[36]、この承認には議論がある[37]。欧州医薬品庁による承認は得られなかった[38]。
EUでは、アタルレンの使用が承認されている[39][40]。
アンチセンスオリゴヌクレオチドゴロディルセンは2019年にアメリカ合衆国で医療用途での使用が承認された。エクソン53スキップ応答症例の治療に有効である[41][42]。
モルフォリノアンチセンスオリゴヌクレオチドビルトラルセンは、2020年夏にアメリカ合衆国で医療用途での使用が承認された[43]。エクソン53スキップ応答変異が確認された患者の治療に利用される[44]。このタイプの変異に対する標的治療としてアメリカ合衆国で2番目に承認された医薬品である[44]。DMD患者の約8%がこの変異を抱えている[44]。
カシメルセンは2021年2月にアメリが合衆国で医療用途での使用が承認された[45]。エクソン45スキップ応答変異が確認された患者に対する標的治療として初めてFDAの承認を受けた医薬品である[45]。
アメリカ疾病予防管理センターによってDMDのケアに対する多職種・包括的なガイドラインが作成され、2010年にThe Lancet Neurologyにおいて2部に分けて発表された[25]。改訂版が2018年に発表されている[46][47]。
理学療法
予後
デュシェンヌ型筋ジストロフィーは進行性の希少疾患であり、最終的には全ての随意筋が影響を受け、心筋や呼吸筋も影響を受ける。平均寿命は25–26歳と推計されているが[3][13]、個人差が大きい。優れた医療ケアが行われた場合、患者は30代まで生存することが多い[48]。この疾患で最高齢の可能性がある人物は、2021年時点で58歳であった[49]。
DMDの患者の直接的な死因として最も一般的なのは呼吸不全である。人工呼吸器や気管切開などの治療の合併症も懸念点となる。次に多い死因は、拡張型心筋症による心不全などの心臓関連疾患である。呼吸補助が行われた場合、生存期間の中央値は40歳にまで伸びる。車いすやベッドを適切に使用し、補助換気、気道クリアランス、心臓症状に対する投薬を行うことで40代もしくは50代前半まで生存する症例も稀にある[50]。今後のケアに関して必要なサポートを早期に計画しておくことが、DMDの患者の寿命の延長につながることが示されている[51]。
デュシェンヌ型筋ジストロフィーのmdxマウスモデルでは、ジストロフィンの欠損がカルシウムレベルの上昇、骨格筋の筋壊死と関係している。また、内喉頭筋(ILM)は保護され、筋壊死が起こらない[52]。ILMは他の筋肉と比較して、カルシウム濃度の変化への対処能力が高いことを示唆するカルシウム調節系プロファイルを示す。このことからはILMの独特な病態生理学的特性の根底にある機構に対する洞察がもたらされる可能性があり[53]、またILMの研究はさまざまな臨床シナリオにおける筋消耗の予防と治療の新規戦略の開発につながる可能性がある[54]。
疫学
DMDは最も一般的なタイプの筋ジストロフィーであり、その発生率は出生男児3600人[13]もしくは5000人[2]に1人と推計されている。
アメリカ合衆国の2010年の研究では、5歳から54歳までのDMDの患者の内訳は非ヒスパニック白人や黒人と比較してヒスパニックが多いことが示されている[55]
研究
 |
筋ジストロフィーはどのタイプも根本的治療法が存在しない[56]。遺伝子治療(マイクロジストロフィン)やアンチセンス治療(エテプリルセンなど)といった、根本原因に対処するよう設計されたいくつかの薬剤が開発中であり[57]、ジストロフィンまたはウトロフィン(ユートロフィン)のいずれかの産生能力を回復する薬剤の探索が行われている[58]。他の取り組みとしては、筋細胞へのカルシウムの流入の遮断の試みなどが行われている[59]。
また、近年の3つの進歩によって、DMDなどの筋ジストロフィーの治療状況は変化する可能性が高い。1つは、人工多能性幹細胞(iPS細胞)を用いた効果的な治療戦略のデザインである。次に、人工知能(AI)は治療標的の発見に役立つ可能性がある。また、疾患モデルを用いて多様なソースから収集された大量のマルチオミックスデータからは、さまざまな経路の収束や分岐に関して有用な情報がもたらされる可能性がある[20]。
エクソンスキッピング
DNAの構造的アナログであるアンチセンスオリゴヌクレオチドは、DMD患者の10%の治療法となる可能性がある[60]。これらの化合物は、ジストロフィン遺伝子がRNAへ転写される際に遺伝子の欠陥のある部分をスキップ(エクソンスキッピング)することで、切り詰められた形ではあるもののより機能的なタンパク質の産生を可能にする[61]。
2種類のアンチセンスオリゴ、2'-O-メチルチオリン酸オリゴ(ドリサペルセン)とモルフォリノオリゴ(エテプリルセンなど)で暫定的なベネフィットのエビデンスが得られており、現在も研究が行われている[62]。エテプリルセンはエクソン51をスキップするよう標的化されている。エクソン51のスキップによって、欠失を抱える男児の約15%でリーディングフレームが回復する。10か所の異なるエクソンを標的とするアンチセンスオリゴによって、欠失を抱える男児の70%以上への対処が可能になることが示唆されている[60]。

DMDよりも軽症であるベッカー型筋ジストロフィーの患者では、ジストロフィンタンパク質は正常よりも短いものの、機能を保持している[63]。1990年Englandらによって、軽症型であるベッカー型筋ジストロフィーの患者がジストロフィン遺伝子のコーディング領域の46%を欠失していることが記載された[63]。こうした切り詰められているものの機能的な形態のジストロフィンの存在から、正常よりも短い形のジストロフィンであっても治療的ベネフィットが得られるという発想が得られた。また同時期にKoleらによって、アンチセンスオリゴヌクレオチドによるpre-mRNAの標的化によるスプライシングの変化が報告された[64]。Koleらはβサラセミアの患者から得られた細胞で、スプライシング過程を標的としたアンチセンスオリゴヌクレオチドによって誤ったスプライシングを修正することに成功した[65][66]。Wiltonのグループによって、筋ジストロフィーへのエクソンスキッピングの応用が試みられた[67][68]。
遺伝子治療
DMDの原因となる変異を修正する、遺伝子編集による治療法の開発へ向けた取り組みが現在行われている[69]。CRISPR/Cas9によるゲノム編集技術によってジストロフィン遺伝子中の変異を正確に除去し、DNA修復機構によって遺伝子の正常なコピーで置き換える、というものである[70][71]。CRISPR/Cas9システムによるゲノム編集は、現在のところヒトで実行可能な状態ではない。しかしながら技術の進歩によって、将来的にはDMDの治療のためにこの技術を用いることができるようになる可能性がある[72][73]。
2007年には、DMDに対するウイルスベクターを用いた遺伝子治療の最初の臨床試験が行われた[74]。DMDやベッカー型筋ジストロフィーに対する遺伝子治療には、Biostrophinと呼ばれるデリバリーベクターが用いられている[75]。
歴史

この疾患はナポリの医師Giovanni Semmolaによって1834年に、そしてGaetano Conteによって1836年に記載された[76][77][78]。しかしながら、デュシェンヌ型筋ジストロフィーの名称はフランスの神経学者ギヨーム=バンジャマン=アマン・デュシェンヌ(1806–1875)に由来している。彼は1861年の著作Paraplegie hypertrophique de l'enfance de cause cerebraleにおいて、この疾患の少年の症例について詳細に記載した。その1年後、彼は自身の患者の写真をAlbum de photographies pathologiquesとして発表した。また、1868年には他の13人の患者について報告した。デュシェンヌは、顕微鏡検査のために存命中の患者から組織を採取する、生検を行った最初の人物である[79][80]。
脚注
出典
- ^ a b c d e f g “NINDS Muscular Dystrophy Information Page”. NINDS (March 4, 2016). 30 July 2016時点のオリジナルよりアーカイブ。12 September 2016閲覧。
- ^ a b c d e f g h i j k l m n o p q r s t u v w x y “Muscular Dystrophy: Hope Through Research”. NINDS (March 4, 2016). 30 September 2016時点のオリジナルよりアーカイブ。12 September 2016閲覧。
- ^ a b c Lisak, Robert P.; Truong, Daniel D.; Carroll, William; Bhidayasiri, Roongroj (2011). International Neurology. Wiley. p. 222. ISBN 9781444317015
- ^ a b c “Phenotypic contrasts of Duchenne Muscular Dystrophy in women: Two case reports.”. Sleep Sci 9 (3): 129–133. (2016). doi:10.1016/j.slsci.2016.07.004. PMC 5241604. PMID 28123647.
- ^ Hamilton, Jon (27 July 2020). “A Boy with Muscular Dystrophy Was Headed for a Wheelchair. Then Gene Therapy Arrived”. NPR
- ^ “Muscular Dystrophy: Hope Through Research”. National Institute of Neurological Disorders and Stroke. 10 August 2020閲覧。
- ^ a b c d e “Duchenne muscular dystrophy | Genetic and Rare Diseases Information Center (GARD) – an NCATS Program”. rarediseases.info.nih.gov. 2021年1月24日閲覧。
- ^ Angelini, C; Tasca, E (December 2012). “Fatigue in muscular dystrophies.”. Neuromuscular Disorders 22 Suppl 3: S214-20. doi:10.1016/j.nmd.2012.10.010. PMC 3526799. PMID 23182642.
- ^ a b Rowland, L.P. (1985). “Clinical Perspective: Phenotypic Expression In Muscular Dystrophy”. In Strohman, C.; Wolf, S.. Gene Expression in Muscle. Advances in Experimental Medicine and Biology. Plenum Press. pp. 3–5. ISBN 978-1-4684-4907-5
- ^ a b c d e f Darras, BT; Urion, DK; Ghosh, PS; Adam, MP; Ardinger, HH; Pagon, RA et al. (2018). Dystrophinopathies. PMID 20301298.
- ^ Emery, Alan E. H.; Muntoni, Francesco; Quinlivan, Rosaline C. M. (2015). Duchenne Muscular Dystrophy (Fourth ed.). OUP Oxford. ISBN 978-0-19968148-8 27 May 2020閲覧。
- ^ a b “Muscular dystrophy - Symptoms and causes”. Mayo Clinic. 2015年2月6日時点のオリジナルよりアーカイブ。2015年2月6日閲覧。
- ^ a b c d MedlinePlus Encyclopedia Duchenne muscular dystrophy
- ^ Sutherland, DH; Olshen, R; Cooper, L; Wyatt, M; Leach, J; Mubarak, S et al. (February 1981). “The pathomechanics of gait in Duchenne muscular dystrophy.”. Developmental Medicine and Child Neurology 23 (1): 3–22. doi:10.1111/j.1469-8749.1981.tb08442.x. PMID 7202868.
- ^ Baptista, CR; Costa, AA; Pizzato, TM; Souza, FB; Mattiello-Sverzut, AC (2014). “Postural alignment in children with Duchenne muscular dystrophy and its relationship with balance.”. Brazilian Journal of Physical Therapy 18 (2): 119–26. doi:10.1590/s1413-35552012005000152. PMC 4183248. PMID 24838810.
- ^ Doorenweerd, N; Mahfouz, A; van Putten, M; Kaliyaperumal, R; T' Hoen, PAC; Hendriksen, JGM et al. (3 October 2017). “Timing and localization of human dystrophin isoform expression provide insights into the cognitive phenotype of Duchenne muscular dystrophy.”. Scientific Reports 7 (1): 12575. Bibcode: 2017NatSR...712575D. doi:10.1038/s41598-017-12981-5. PMC 5626779. PMID 28974727.
- ^ “Morphofunctional aspects of the blood-brain barrier”. Current Drug Metabolism 13 (1): 50–60. (January 2012). doi:10.2174/138920012798356970. PMID 22292807.
- ^ “High prevalence of plasma lipid abnormalities in human and canine Duchenne and Becker muscular dystrophies depicts a new type of primary genetic dyslipidemia”. J Clin Lipidol 14 (4): 459–69. (July 2020). doi:10.1016/j.jacl.2020.05.098. PMC 7492428. PMID 32593511.
- ^ Online 'Mendelian Inheritance in Man' (OMIM) Muscular Dystrophy, Duchenne Type; DMD -310200
- ^ a b Vera, Carlos D.; Zhang, Angela; Pang, Paul D.; Wu, Joseph C. (2022). “Treating Duchenne Muscular Dystrophy: The Promise of Stem Cells, Artificial Intelligence, and Multi-Omics”. Frontiers in Cardiovascular Medicine 9: 851491. doi:10.3389/fcvm.2022.851491. ISSN 2297-055X. PMC 8960141. PMID 35360042.
- ^ a b “Duchenne Muscular Dystrophy: Pathophysiological Implications of Mitochondrial Calcium Signaling and ROS Production” (2012年5月2日). May 2, 2012時点のオリジナルよりアーカイブ。2014年6月29日閲覧。
- ^ Wahl, Margaret (October 21, 2016). “Quest - Article - But Girls Don't Get Duchenne, or Do They? - A Quest Article”. Muscular Dystrophy Association. July 6, 2019閲覧。
- ^ “Understanding Genetics” (26 November 2021). 2022年9月17日閲覧。
- ^ “University of Utah Muscular Dystrophy”. Genome.utah.edu (2009年11月28日). 2013年2月16日閲覧。
- ^ a b “Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and pharmacological and psychosocial management”. The Lancet. Neurology 9 (1): 77–93. (January 2010). doi:10.1016/s1474-4422(09)70271-6. PMID 19945913.
- ^ “Integrated study of 100 patients with Xp21 linked muscular dystrophy using clinical, genetic, immunochemical, and histopathological data. Part 2. Correlations within individual patients”. Journal of Medical Genetics 30 (9): 737–44. (September 1993). doi:10.1136/jmg.30.9.737. PMC 1016530. PMID 8411068.
- ^ “Is a muscle biopsy in Duchenne dystrophy really necessary?”. Neurology 57 (4): 574–5. (August 2001). doi:10.1212/wnl.57.4.574. PMID 11524463.
- ^ “Rapid direct sequence analysis of the dystrophin gene”. American Journal of Human Genetics 72 (4): 931–9. (April 2003). doi:10.1086/374176. PMC 1180355. PMID 12632325.
- ^ Beksac, Mehmet Sinan; Tanacan, Atakan; Aydin Hakli, Duygu; Orgul, Gokcen; Soyak, Burcu; Balci Hayta, Burcu et al. (30 July 2018). “Gestational Outcomes of Pregnant Women Who Have Had Invasive Prenatal Testing for the Prenatal Diagnosis of Duchenne Muscular Dystrophy”. Journal of Pregnancy 2018: 1–5. doi:10.1155/2018/9718316. PMC 6091284. PMID 30151283.
- ^ Devaney, Stephanie A.; Palomaki, Glenn E.; Scott, Joan A.; Bianchi, Diana W. (2011-08-10). “Noninvasive Fetal Sex Determination Using Cell-Free Fetal DNA: A Systematic Review and Meta-analysis”. JAMA 306 (6): 627–636. doi:10.1001/jama.2011.1114. ISSN 0098-7484. PMC 4526182. PMID 21828326.
- ^ “Duchenne Muscular Dystrophy Statement”. Drug Safety and Availability. US FDA (2014年10月31日). 2014年11月2日時点のオリジナルよりアーカイブ。2022年9月18日閲覧。
- ^ “Pfizer's New Phase 1b Results of Gene Therapy in Ambulatory Boys with Duchenne Muscular Dystrophy (DMD) Support Advancement into Pivotal Phase 3 Study | Pfizer”. 2022年9月23日閲覧。
- ^ “Construction of a Quality of Life Questionnaire for slowly progressive neuromuscular disease”. Quality of Life Research 24 (11): 2615–23. (November 2015). doi:10.1007/s11136-015-1013-8. PMID 26141500.
- ^ “Corticosteroids for the treatment of Duchenne muscular dystrophy”. The Cochrane Database of Systematic Reviews 5 (5): CD003725. (May 2016). doi:10.1002/14651858.CD003725.pub4. PMC 8580515. PMID 27149418.
- ^ “Cardiac involvement in patients with muscular dystrophies: magnetic resonance imaging phenotype and genotypic considerations”. Circulation: Cardiovascular Imaging 4 (1): 67–76. (January 2011). doi:10.1161/CIRCIMAGING.110.960740. PMC 3057042. PMID 21245364.
- ^ “FDAetep”. Food and Drug Administration (19 September 2016). 8 July 2019閲覧。
- ^ “Railroading at the FDA”. Nature Biotechnology 34 (11): 1078. (2016-11-08). doi:10.1038/nbt.3733. PMID 27824847.
- ^ “CHMP Advises Against Approval for Eteplirsen in DMD”. Medscape. 2019年7月9日閲覧。
- ^ “Translarna EPAR”. European Medicines Agency (EMA). 14 August 2020閲覧。
- ^ “Translarna - Summary of Product Characteristics (SmPC)”. (emc) (24 April 2017). 18 June 2017閲覧。
- ^
"FDA grants accelerated approval to first targeted treatment for rare Duchenne muscular dystrophy mutation". U.S. Food and Drug Administration (FDA) (Press release). 12 December 2019. 2019年12月13日時点のオリジナルよりアーカイブ。2019年12月12日閲覧。
 この記述には、アメリカ合衆国内でパブリックドメインとなっている記述を含む。
この記述には、アメリカ合衆国内でパブリックドメインとなっている記述を含む。 - ^ “Drug Approval Package: Vyondys 53 (golodirsen)”. U.S. Food and Drug Administration (FDA) (21 January 2020). 22 January 2020閲覧。
- ^ Roshmi, Rohini Roy; Yokota, Toshifumi (2023). “Viltolarsen: From Preclinical Studies to FDA Approval”. Methods in Molecular Biology (Clifton, N.J.) 2587: 31–41. doi:10.1007/978-1-0716-2772-3_2. ISSN 1940-6029. PMID 36401022.
- ^ a b c
"FDA Approves Targeted Treatment for Rare Duchenne Muscular Dystrophy Mutation". U.S. Food and Drug Administration (FDA) (Press release). 12 August 2020. 2020年8月12日閲覧。 この記述には、アメリカ合衆国内でパブリックドメインとなっている記述を含む。
- ^ a b
"FDA Approves Targeted Treatment for Rare Duchenne Muscular Dystrophy Mutation". U.S. Food and Drug Administration (FDA) (Press release). 25 February 2021. 2021年2月25日閲覧。 この記述には、アメリカ合衆国内でパブリックドメインとなっている記述を含む。
- ^ “Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and neuromuscular, rehabilitation, endocrine, and gastrointestinal and nutritional management”. Lancet Neurol 17 (3): 251–267. (March 2018). doi:10.1016/S1474-4422(18)30024-3. PMC 5869704. PMID 29395989.
- ^ “Diagnosis and management of Duchenne muscular dystrophy, part 2: respiratory, cardiac, bone health, and orthopaedic management”. Lancet Neurol 17 (4): 347–361. (April 2018). doi:10.1016/S1474-4422(18)30025-5. PMC 5889091. PMID 29395990.
- ^ “Duchenne muscular dystrophy (DMD) | Muscular Dystrophy Campaign”. Muscular-dystrophy.org. 2013年1月21日時点のオリジナルよりアーカイブ。2013年2月16日閲覧。
- ^ Carter, Nicole (January 14, 2021). “Nursing home resident defies COVID, wants to eat out again”. Sun Journal
- ^ “Evolution of life expectancy of patients with Duchenne muscular dystrophy at AFM Yolaine de Kepper centre between 1981 and 2011”. Annals of Physical and Rehabilitation Medicine 56 (6): 443–54. (September 2013). doi:10.1016/j.rehab.2013.06.002. PMID 23876223.
- ^ “[Personal experimental experience with the administration of liquid obliterative agents using percutaneous intra-arterial balloon catheters with a controlled leak”]. Sbornik Vedeckych Praci Lekarske Fakulty Karlovy Univerzity V Hradci Kralove. Supplementum 30 (2): 201–11. (2012-11-22). doi:10.1186/1750-1172-7-S2-A8. PMC 3504593. PMID 3504593.
- ^ “Intrinsic laryngeal muscles are spared from myonecrosis in the mdx mouse model of Duchenne muscular dystrophy”. Muscle & Nerve 35 (3): 349–53. (March 2007). doi:10.1002/mus.20697. PMID 17143878.
- ^ “Expression of calcium-buffering proteins in rat intrinsic laryngeal muscles”. Physiological Reports 3 (6): e12409. (June 2015). doi:10.14814/phy2.12409. PMC 4510619. PMID 26109185.
- ^ “Intrinsic Laryngeal Muscles and Potential Treatments for Skeletal Muscle-Wasting Disorders”. Austin Journal of Otolaryngology 1 (1): 3. (2014). オリジナルの2015-06-26時点におけるアーカイブ。.
- ^ “Key Findings: Prevalence of Duchenne / Becker Muscular Dystrophies” (英語). Centers for Disease Control and Prevention. (2018年1月5日) 2018年11月18日閲覧。
- ^ “Muscular Dystrophy Information Page: National Institute of Neurological Disorders and Stroke (NINDS)”. www.ninds.nih.gov. 30 July 2016時点のオリジナルよりアーカイブ。22 February 2022閲覧。
- ^ “Muscular Dystrophy: Hope Through Research”. www.ninds.nih.gov. 30 September 2016時点のオリジナルよりアーカイブ。22 February 2022閲覧。
- ^ “Pharmacological advances for treatment in Duchenne muscular dystrophy”. Current Opinion in Pharmacology 34: 36–48. (June 2017). doi:10.1016/j.coph.2017.04.002. PMID 28486179.
- ^ “Calcium and the damage pathways in muscular dystrophy”. Canadian Journal of Physiology and Pharmacology 88 (2): 83–91. (February 2010). doi:10.1139/Y09-058. PMID 20237582.
- ^ a b “Genetic therapies for inherited neuromuscular disorders”. The Lancet. Child & Adolescent Health 2 (8): 600–609. (August 2018). doi:10.1016/S2352-4642(18)30140-8. PMID 30119719.
- ^ “Modification of splicing in the dystrophin gene in cultured Mdx muscle cells by antisense oligoribonucleotides”. Human Molecular Genetics 7 (7): 1083–90. (July 1998). doi:10.1093/hmg/7.7.1083. PMID 9618164.
- ^ "FDA grants accelerated approval to first drug for Duchenne muscular dystrophy" (Press release). FDA. 19 September 2016. 2016年12月11日時点のオリジナルよりアーカイブ。2016年12月12日閲覧。
- ^ a b “Very mild muscular dystrophy associated with the deletion of 46% of dystrophin”. Nature 343 (6254): 180–2. (January 1990). Bibcode: 1990Natur.343..180E. doi:10.1038/343180a0. PMID 2404210.
- ^ “Restoration of correct splicing in thalassemic pre-mRNA by antisense oligonucleotides”. Proceedings of the National Academy of Sciences of the United States of America 90 (18): 8673–7. (September 1993). Bibcode: 1993PNAS...90.8673D. doi:10.1073/pnas.90.18.8673. PMC 47420. PMID 8378346.
- ^ “Restoration of hemoglobin A synthesis in erythroid cells from peripheral blood of thalassemic patients”. Proceedings of the National Academy of Sciences of the United States of America 97 (17): 9591–6. (August 2000). Bibcode: 2000PNAS...97.9591L. doi:10.1073/pnas.97.17.9591. PMC 16909. PMID 10944225.
- ^ “Restoration of human beta-globin gene expression in murine and human IVS2-654 thalassemic erythroid cells by free uptake of antisense oligonucleotides”. Molecular Pharmacology 62 (3): 545–53. (September 2002). doi:10.1124/mol.62.3.545. PMID 12181431.
- ^ “Specific removal of the nonsense mutation from the mdx dystrophin mRNA using antisense oligonucleotides”. Neuromuscular Disorders 9 (5): 330–8. (July 1999). doi:10.1016/S0960-8966(99)00010-3. PMID 10407856.
- ^ “Antisense oligonucleotide-induced exon skipping across the human dystrophin gene transcript”. Molecular Therapy 15 (7): 1288–96. (July 2007). doi:10.1038/sj.mt.6300095. PMID 17285139.
- ^ “Correction of diverse muscular dystrophy mutations in human engineered heart muscle by single-site genome editing”. Science Advances 4 (1): eaap9004. (January 2018). Bibcode: 2018SciA....4P9004L. doi:10.1126/sciadv.aap9004. PMC 5796795. PMID 29404407.
- ^ Cohen, Jon (2018-08-30). “Gene editing of dogs offers hope for treating human muscular dystrophy”. Science. doi:10.1126/science.aav2676.
- ^ Patmanathan, Sathya Narayanan; Gnanasegaran, Nareshwaran; Lim, Moon Nian; Husaini, Roslina; Fakiruddin, Kamal Shaik; Zakaria, Zubaidah (2018). “CRISPR/Cas9 in Stem Cell Research: Current Application and Future Perspective”. Current Stem Cell Research & Therapy 13 (8): 632–644. doi:10.2174/1574888X13666180613081443. PMID 29895256.
- ^ “Prevention of muscular dystrophy in mice by CRISPR/Cas9-mediated editing of germline DNA”. Science 345 (6201): 1184–1188. (September 2014). Bibcode: 2014Sci...345.1184L. doi:10.1126/science.1254445. PMC 4398027. PMID 25123483.
- ^ Wade, Nicholas (31 December 2015). “Gene Editing Offers Hope for Treating Duchenne Muscular Dystrophy, Studies Find”. The New York Times. オリジナルの2 January 2016時点におけるアーカイブ。 1 January 2016閲覧。
- ^ “Gene therapy for duchenne muscular dystrophy: expectations and challenges”. Archives of Neurology 64 (9): 1236–41. (September 2007). doi:10.1001/archneur.64.9.1236. PMID 17846262.
- ^ “Chronicles in drug discovery”. Drug News & Perspectives 18 (8): 517–22. (October 2005). doi:10.1358/dnp.2005.18.8.953409. PMID 16391721.
- ^ Politano, Luisa. “Cardiomiologia e Genetica Medica” [Cardiomyology and Medical Genetics] (イタリア語). Seconda Università degli Studi di Napoli. July 4, 2015時点のオリジナルよりアーカイブ。August 24, 2015閲覧。
- ^ “Da Conte a Duchenne” [By Conte in Duchenne] (イタリア語). DM. Unione Italiana Lotta alla Distrofia Muscolare (October 2005). March 4, 2016時点のオリジナルよりアーカイブ。August 24, 2015閲覧。
- ^ “One-hundred-seventy-five years of Neapolitan contributions to the fight against the muscular diseases”. Acta Myologica 29 (3): 369–91. (December 2010). PMC 3146338. PMID 21574522.
- ^ “Duchenne muscular dystrophy”. Medterms.com (2011年4月27日). 2012年8月6日時点のオリジナルよりアーカイブ。2013年2月16日閲覧。
- ^ Duchenne de Boulogne - Who Named It?
関連文献
- “Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and neuromuscular, rehabilitation, endocrine, and gastrointestinal and nutritional management”. Lancet Neurol 17 (3): 251–267. (March 2018). doi:10.1016/S1474-4422(18)30024-3. PMC 5869704. PMID 29395989.
- “Diagnosis and management of Duchenne muscular dystrophy, part 2: respiratory, cardiac, bone health, and orthopaedic management”. Lancet Neurol 17 (4): 347–361. (April 2018). doi:10.1016/S1474-4422(18)30025-5. PMC 5889091. PMID 29395990.
外部リンク
- DMDを知る -デュシェンヌ型筋ジストロフィーの専門情報サイト-
- Muscular Dystrophies - Curlie
- CDC's National Center on Birth Defects and Developmental Disabilities (previously listed below as "Duchenne/Becker Muscular Dystrophy, NCBDDD") at CDC
- Genes and Disease Page at NCBI

デュシェンヌ型筋ジストロフィー
出典: フリー百科事典『ウィキペディア(Wikipedia)』 (2018/07/04 17:24 UTC 版)
「イデベノン」の記事における「デュシェンヌ型筋ジストロフィー」の解説
マウスで実験的に確かめられた後、ヒトでの予備的試験が実施され、2005年に第II相臨床試験が、2009年に偽薬対照第III相臨床試験(DELOS試験)が実施された。DELOS試験の結果、最大呼気流量(PEF)の予測値に対する割合(PEF%p)の26週、39週、52週におけるベースラインからの変化量はそれぞれ有意であった(P=0.007、P=0.034、P=0.0001)。52週でのPEF%p変化量はイデベノン群:-3.05%p、偽薬群:-9.01%pであった。
※この「デュシェンヌ型筋ジストロフィー」の解説は、「イデベノン」の解説の一部です。
「デュシェンヌ型筋ジストロフィー」を含む「イデベノン」の記事については、「イデベノン」の概要を参照ください。
デュシェンヌ型筋ジストロフィーと同じ種類の言葉
- デュシェンヌ型筋ジストロフィーのページへのリンク
