きん‐ジストロフィー【筋ジストロフィー】
筋ジストロフィ症
筋ジストロフィー(進行性筋ジストロフィー) ( myotonic dystrophy )
筋ジストロフィー
筋ジストロフィーとは「筋線維の変性・壊死を主病変とし、進行性の筋力低下をみる遺伝子の疾患である」と定義されています。多くは遺伝しますので、その遺伝形式により、分類が行われてきました。しかし、近年分子遺伝学の進歩により、遺伝子座が次々と明らかにされ、またクローニングされ、それに基づいた疾患分類が試みられるようになっています。主な病気の分類を表1に示しました。
| 表1 主な筋ジストロフィー | ||
| × | 遺伝子座 | 遺伝子産物 |
| 1)X連鎖劣性遺伝 | ||
| a. Duchenne型 | Xp21 | dystrophin |
| b. Becker型 | Xp21 | dystrophin |
| c. Emery-Dreifuss型 | Xq28 | emerin |
| 2)常染色体劣性遺伝 | ||
| a. 肢帯型 | (表2) | |
| b. 先天性 | × | × |
| 福山型 | 9q31 | fukutin |
| 非福山型(古典型) | ||
| メロシン欠損型 | 6q | merosin (laminin α2鎖) |
| メロシン陽性型 | × | × |
| c. 遠位型(三好) | 2p13 | dysferlin |
| 3)常染色体優性遺伝 | ||
| a. 顔面肩甲上腕型 | 4q-ter | × |
| b. 肢帯型 | (表2) | |
| c. 眼・咽頭型 | 14q11.2-q13 | poly A binding protein 2 |
A.デュシェンヌ型筋ジストロフィー(Duchenne muscular dystrophy: DMD)
a.病因、病態、病理
本症はX連鎖(性染色体)劣性遺伝をとるため、患者は男児に限られます。同じ様な遺伝形式をとる病気に血友病や色盲があります。母親が遺伝子に異常(変異)をもっていて、それが男の子に伝わることが多いのです。しかし、母親が遺伝子変異をもっているとは限りません。この病気は突然変異率が高いので、お子さんの遺伝子に突然変異があり、お母さんには変異がないことがまれではありません。お母さんの約1/3は変異がありません。
まれに染色体異常(X染色体と常染色体の相互転座、Turner症候群)があると、女性も同様な症状をとります。また女性保因者(女性で遺伝子の異常を持っている人)も、まれですが血液のCK値が高かったり、軽い筋肉の力が弱かったりします。
この病気を起こす遺伝子はどこにあって、その遺伝子に変異があるとどのような蛋白が欠損するか明らかになっています。遺伝子のある場所はX染色体短腕(Xp21)にあります。この遺伝子は分子量427kDの蛋白(ジストロフィンと命名されています)をコードしています。ジストロフィン遺伝子はクローニングされ、それはcDNAで14kbあって、79のエクソンからなっています。患者さんではこのジストロフィン遺伝子に変異があり、ジストロフィン蛋白が生成されないのです。患者さんの50ー60%はジストロフィkン遺伝子の欠失(遺伝子が欠けていて短い)、約10%は重複(遺伝子が重なって長くなっている)です。残りの30ー40%は多分点変異(DNAの一個が間違っているような一塩基置換など)と考えられています。 最も多い欠失は、3塩基づつの読みにずれがある(out of frame)欠失(正常では塩基3個で一つのアミノ酸ができます。3個の組み合わせがずれるので、めちゃくちゃなアミノ酸ができるのです)です。それだけでなく、欠失部以下にもう蛋白を作るのを止めなさいというストップ命令(コドン)が働き、不完全な蛋白が生成されます。それは不安定で、すぐに分解されると考えられています。ですからデュシェンヌ型ではジストロフィン蛋白はまったく作られません。
ジストロフィン蛋白は筋細胞膜直下に局在していて、ジストロフィン結合蛋白と結合しています。ジストロフィンは細長い棒状構造をしていると考えられています(図6)。
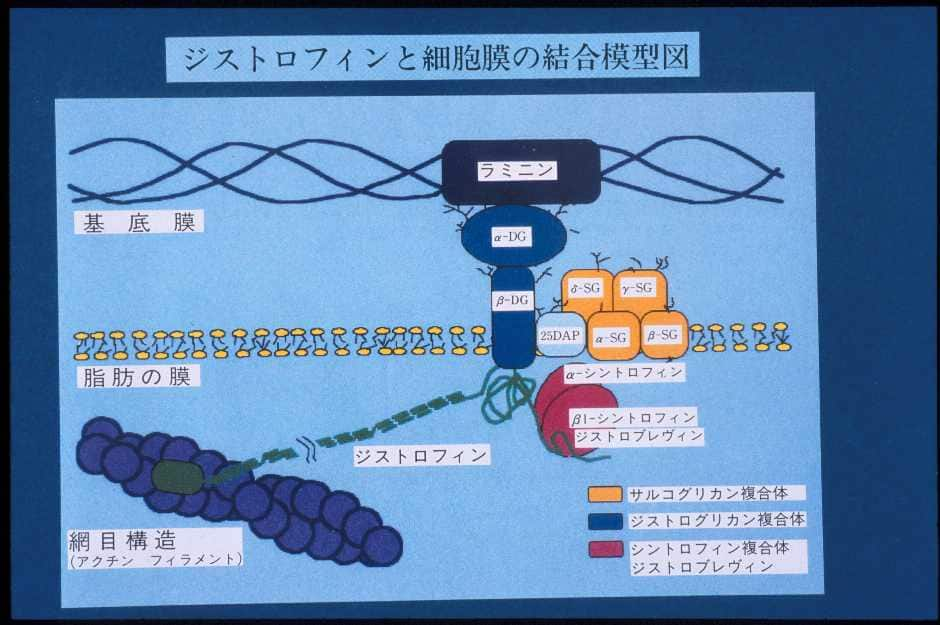 |
| 図6:ジストロフィンとジストロフィン結合蛋白の関係 この図は筋細胞膜(基底膜と形質膜の2重膜からなる)の一部を拡大した分子モデルの図である。 20DAPはサルコスパンとよばれている。DG:ジストログリカン、SG:サルコグリカン |
5’側(ジストロフィンの頭の部分)は細胞膜をしっかりと強固に保つ、アクチンという細い糸と結合する部位(actin binding domain)で、それに続いて3重らせん構造をとる桿状部分(rod domain)が続いています。それはさらにシスティンに富む部分(cysteine-rich domain)、C端(carboxyl terminal)へと続いています。図6のようにジストロフィン分子はcysteine-rich domainのところでジストロフィン結合蛋白のβジストログリカン(β-dystroglycan)と結合しています。ジストロフィンはアクチンという細い線維と一緒になって膜を補強するものです(細胞膜をしっかりと裏打ち構造をするもので細胞骨格といいます)。
患者さんではこのジストロフィンは全く欠損しています。それは抗ジストロフィン抗体で免疫染色をすると、正常では筋細胞膜に局在するジストロフィンが全く染色されないことから容易に判定でき(図7)、本症の診断用に使用されています。細胞膜をしっかりと安定させているジストロフィン蛋白がないので、細胞膜は弱く、すぐにシャボン玉のようにこわれるのでしょう。
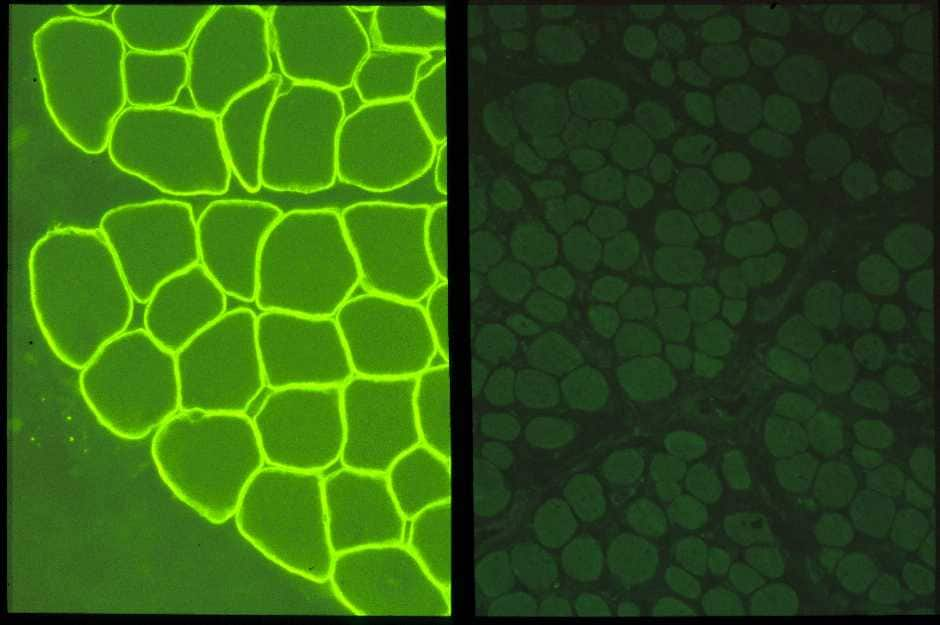 | 正常筋(左)では筋細胞膜にジストロフィンが存在するので、膜の部分が蛍光を発し、染色されている。 デュシェンヌ型(右)ではジストロフィンがないので、まったく染色されていない。 |
| 図7:ジストロフィン抗体による免疫染色 | |
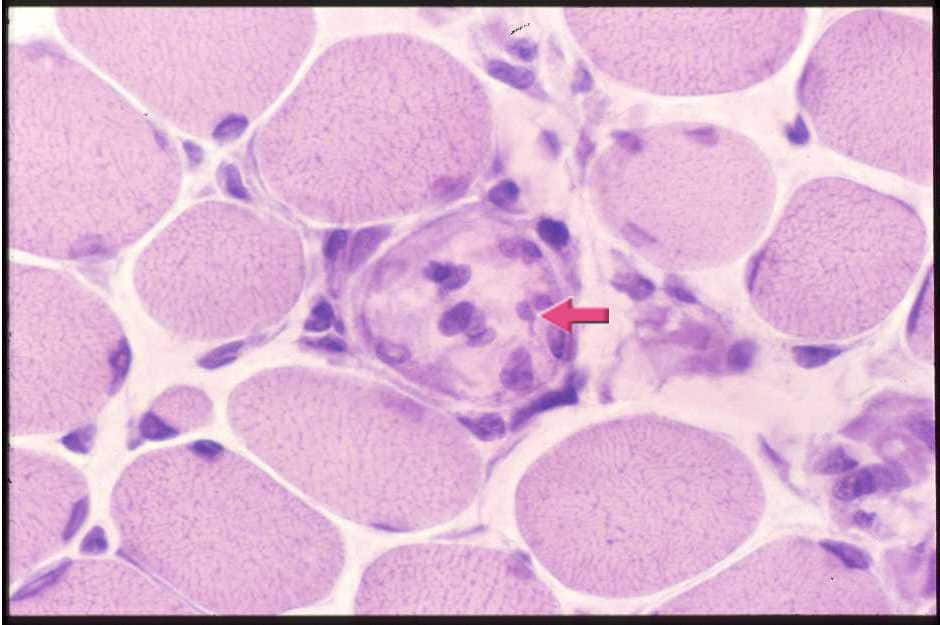 | 筋線維の大小不同と壊死線維(この図の中央にあって、溶けたような胞体をもっている)(矢印)をみる。 壊死線維には大型の核をもつマクロファージが侵入している。 |
| 図8:デュシェンヌ型筋ジストロフィーの筋病理-1 | |
| 筋ジストロフィーにみられる沢山の再生線維。中央にみえる青みを帯びた小さな線維(→) で、デュシェンヌ型では筋線維の平均15%は再生途上筋である。 マクロファージで掃除され、きれいになった壊死細胞には続いて再生が起こる。病気が進むと筋線維は著明に減少し、結合織と脂肪織で置換されてしまう。こうなると力は弱く、関節が硬くなり、伸びなくなってしまう。 | 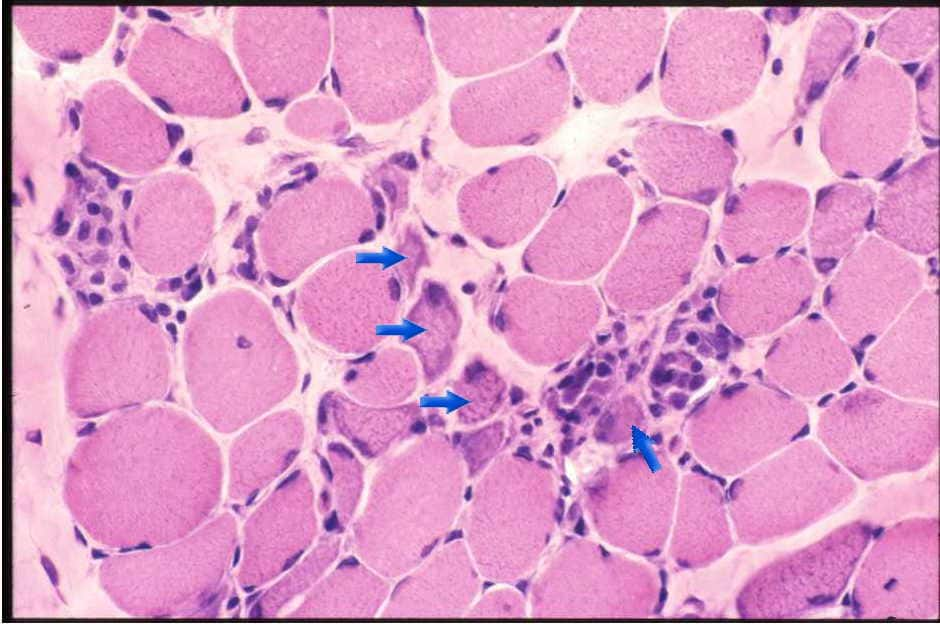 |
| 図9:デュシェンヌ型筋ジストロフィーの筋病理-2 | |
b 本症の頻度は男児出生3、300人に1人、人口10万人に3ー5人くらいの患者さんがいるといわれています。人種差はなく、あらあゆる国に患者さんがいます。
従来は3ー5歳頃、走れない、転びやすい、階段の昇降困難で気付かれることが多かったのです。しかし最近では、乳児期に他の疾患(たとえば風邪をひいたとか)での検査中に偶然高CK血症で見いだされることが多くなりましたた。乳児期から追跡すると歩行開始遅延(1歳6カ月以降に歩行開始)が30ー50%いること、歩行開始時にはすでに筋力低下(立ち上がり方の異常)がみられることから、乳児期にもすでに異常があるお子さんがいるようです。
病初期は上記のように転びやすい、走れないなど歩行に関する異常が最も多くみられます。また筋肉の力が明らかに弱くなると、そん居の姿勢から立ち上がるとき、床に手をつき、臀部を高く挙げて立つようになります。健康なお子さんは、しゃがんだ位置からすっと垂直に立ち上がります。さらに筋肉の力が弱くなると、手を床に着き、つぎに膝に手を交互にあてて立つ、いわゆる登はん性起立(Gowers徴候)をみるようになります
(図10)。
| 腰の筋力低下があるため、床から起立する時、まず床に手をついて、お尻を高くあげる(a)。 次にひざに手をあてて、手の力を借りて立ち上がる(b)。 ふくらはぎは太く偽性肥大を示している。 この偽性肥大はデュシェンヌ型、ベッカー型に特徴的である。 | 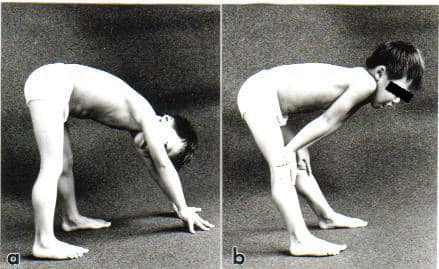 |
| 図10: 登はん性起立(Gowers徴候) | |
さらに進行すると、何かものにつかまらないと立てなくなり、10歳前後で歩行不能となり、車椅子生活となります。次第に寝返りにも人の介助が必要となり、20歳前後で、呼吸筋の力が弱くなるため、人工呼吸器の助けが必要となります。むかしは人工呼吸器がなかったので、患者さんは20歳以前に死亡していました。でも今は40歳まで生きる方がでてきました。医療機器の進歩で、患者さんの生命はもっともっと伸びるでしょう。
筋萎縮(見た目に筋肉が細くなる)は病初期にはあまり目立たちません。むしろふくらはぎが異常に太いのが特徴的で、これは仮(偽)性肥大(pseudohypertrophy)とよばれています。この筋の肥大は肩や、頬筋、舌筋にもみられます。ふくらはぎの肥大はデュシェンヌ型や次に述べるベッカー型では、ほとんどの患者さんが経験する病気に特徴的所見です。
病気が進行すると、筋萎縮は躯幹近位筋(大腿、上腕、躯幹筋)に著明にみられるようになります。歩行時には関節拘縮(関節の伸展が悪くなることです)はアキレス腱の短縮による尖足のみですが、歩行不能となった時点から、股関節、膝関節などに広がっていきます。脊柱変形、手指、顎関節など全身の関節の拘縮をみるようになるのです。腱反射はアキレス腱反射を除いて減弱ないし消失します(図11)。
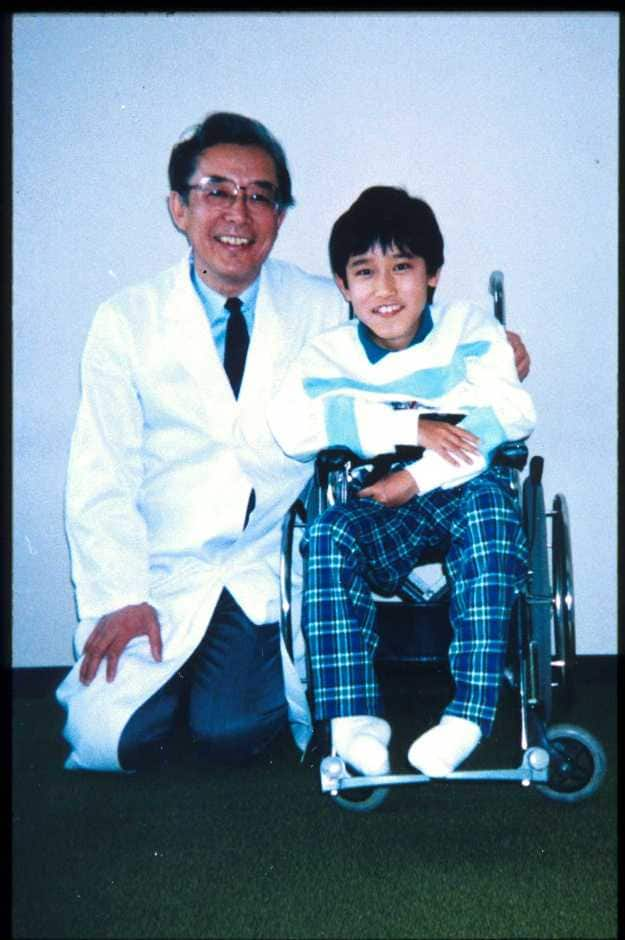 | 足の関節が拘縮している。 T君は今人工呼吸器をつけているが、亜細亜大学法学部に通学し、勉強しているファイトあふれる青年(この写真の掲載はT君の許可を得たものです)。 |
| 図11:車イス生活になったデュシェンヌ型筋ジストロフィーのT君と筆者 | |
現在のところ病気の進行をとめたり、筋力が回復するような根本的治療法はみつかっていません。でも、病気の進行を遅らせるさまざまな試みがなされています。
その一つとして副腎皮質ホルモン投与が試みられています。副作用が出ない少量投与でよい結果が出ています。 わたしはデュボビッツ(Dubowitz)先生の方法(毎月10日間だけプレドニゾロン0.75mg/Kg/日投与、20日間休薬)をとっています。 これですと副作用が出る人はごくまれです。副作用もほっぺがふっくらとする程度です。 中にはとても効果があって、5年くらい病気の進行がなくなる人もいます。 でも、効果がはっきりしない人もいます。アメリカの調査では副腎皮質ホルモン治療を受けた人は歩行期間が2年も延長するとのことです。
最近、アメリカの神経学会、小児神経学会では多くの患者で治験をしたところ、最初はプレドニソン0.75mg/Kg/日の連日投与から開始することがもっとも有効であったとして、推薦しています。副作用が出てきたら、0.3mg/Kg/日に減量します。(http://www.aan.com/professionals/practice/pdfs/DMD_Guideline_Physicians.pdf)。 この量ですと、日本人には量が多すぎて、副作用が出る可能性が強いと思います。日本人用のスタンダードが欲しいです。
デュシェンヌ型では筋力低下、関節拘縮が進行するので、それを少しでも防ぐためのリハビリテーションが行われ、効果をあげています。 また呼吸不全をみるようになった場合は鼻マスクによる人工呼吸器(nasal:noninvasive intermittent positive pressure: NIPP)の使用、あるいは気管切開による人工呼吸器の使用が行われ、患者さんの延命効果に大きく寄与しています。 心不全徴候がみられるようになった患者さんには、アンジオテンシン変換酵素阻害剤(ACE阻害剤)、ベーターブロッカー、ジギタリス剤、利尿剤を組み合わせた治療が行われていて、とてもよい結果をえています。 いろいろな薬物治療、呼吸管理などで、患者さんの生命予後は飛躍的によくなっています。
遺伝子治療はmdxマウス(ジストロフィンが欠損している筋ジストロフィーマウス)を使用して行われています。 現在アデノウィルスDNAをベクター(DNAの細胞内への運びやさん)にして、ジストロフィン遺伝子の一部あるいは全長が挿入され、筋内投与が行われています。 投与部にはジストロフィン陽性線維が増加し、筋線維の壊死の抑制がみられています。ただし、1回投与での効果の持続性が短く、再投与での効果に乏しいことが欠点とされています。 でもアデノウィルスベクターに変わるベクター(たとえばアデノウィルス随伴ウィルスベクター)なども開発されています。 遺伝子治療はよいベクターの開発にかかっています。科学の進歩はすばらしいです。原因がまったく分からなかったデュシェンヌ型筋ジストロフィーの遺伝子がクローニングされたのが1986年です。 最近の学問の進歩には目を見張るものがあります。根本治療、遺伝子治療への時代に突入したのです。
遺伝子治療については別に、国立精神・神経センター神経研究所 遺伝子治療研究部 武田伸一部長に解説いただいています。
→デュシェンヌ型筋ジストロフィーに対する分子治療のこころみ
筋ジストロフィー
出典: フリー百科事典『ウィキペディア(Wikipedia)』 (2024/10/06 05:08 UTC 版)
 | この記事は検証可能な参考文献や出典が全く示されていないか、不十分です。 (2011年10月) |
| 筋ジストロフィー | |
|---|---|
| 概要 | |
| 診療科 | 神経学, 小児科学, 遺伝医学 |
| 分類および外部参照情報 | |
| ICD-10 | G71.0 |
| ICD-9-CM | 359.0-359.1 |
筋ジストロフィー(きんジストロフィー、英語: muscular dystrophy)とは、筋線維の破壊・変性(筋壊死)と再生を繰り返しながら、次第に筋萎縮と筋力低下が進行していく遺伝性筋疾患の総称である。発症年齢や遺伝形式、臨床的経過などからさまざまな病型に分類される。そのうち、最も頻度の高いのはデュシェンヌ型である。2015年7月に難病指定され、日本国内の患者数は約25,400人と推計されている。
定義
主訴が筋力低下、筋萎縮であり、以下の2項目を満たすものをいう。
- 遺伝性疾患である。
- 骨格筋がジストロフィー変化を示す。
ジストロフィー変化とは、筋線維の大小不同、円形化、中心核の増加、結合組織の増生、脂肪化を特徴として筋線維束の構造が失われる変化のことをいう。これは筋ジストロフィーの中で最初に報告されたデュシェンヌ型の病理所見から定義されたものである。
特別支援教育における罹患児の教育については、肢体不自由ないしは病弱で対応する。
筋ジストロフィー (Muscular Dystrophy, MD)
性染色体劣性遺伝型筋ジストロフィー
- デュシェンヌ型 (Duchenne muscular dystrophy, DMD)
- 進行性筋ジストロフィーの大部分を占め、重症な型である。おおよそ小学校5年生くらいの10歳代で車椅子生活となる人が多い。昔は20歳前後で心不全・呼吸不全のため死亡するといわれていたが、「侵襲的人工呼吸法」(気管切開を用いる)や最近では「非侵襲的人工呼吸法」(気管切開などの方法を用いない)など医療技術の進歩により、5年から10年は生命予後が延びている。しかし、未だ根本的な治療法が確立していない難病である。このデュシェンヌ型は、伴性劣性遺伝(X染色体短腕のジストロフィン遺伝子欠損)で基本的に男性のみに発病する。
- 症状
- 2 - 5歳ごろから歩き方がおかしい、転びやすいなどの症状で発症が確認されることが多数である。初期には腰帯筋、次第に大殿筋、肩甲帯筋へと筋力の低下の範囲を広げていく。なお、筋力低下は対称的に起きるという特徴を持つ。また、各筋の筋力低下によって処女歩行遅滞、易転倒、登攀性起立(とうはんせいきりつ、ガワーズ(Gowers)兆候)、腰椎の前弯強、動揺性歩行(アヒル歩行)[注釈 1]などをきたす。筋偽牲肥大に関しては腓腹筋や三角筋で特徴的に起こるが、これは筋組織の崩壊した後に脂肪組織が置き換わることによる仮性肥大である。病勢の進行と共に筋の萎縮(近位→遠位)に関節拘縮、アキレス腱の短縮なども加わり、起立・歩行不能となる。心筋疾患を合併することが多く、心不全は大きな死因のひとつである。
- 検査
- 血清CK値著明に上昇。筋電図にて筋原性変化を認める。尿中クレアチニン↓。尿中クレアチン↑。筋生検にて免疫染色を行いジストロフィン蛋白欠損。
- 治療
- 現在のところ、根本的治療法はない。機能訓練や関節拘縮予防のためのストレッチ(理学療法)のほか、心不全・呼吸障害に対する対症療法が行われる。作用機序は明らかではないが、プレドニゾロンはDMD型筋ジストロフィーに保険適用がある。国産初のアンチセンス核酸医薬品として治療剤(ビルトラルセン)の臨床試験が開始され[1]、2020年3月25日に承認された[2]。また、ヒストン脱アセチル化酵素阻害剤(HDAC阻害剤)による治療の研究がされている。
- ベッカー型 (Becker muscular dystrophy, BMD)
- 病態はデュシェンヌ型と同じだが、発症時期が遅く、症状の進行も緩徐。関節拘縮も少ない。一般に予後は良い。
- デュシェンヌ型同様、免疫染色にてジストロフィン蛋白に異常を認めるが、デュシェンヌ型ではジストロフィン蛋白がほとんど発現していないのに対し、ベッカー型では異常なジストロフィン蛋白が産生されたり、発現量が少ないことが知られており、これにより両者の症状の差異が生じているのだと考えられる。
デュシェンヌ型筋ジストロフィーの筋病理
筋ジストロフィーの筋病理の主要所見は筋線維の壊死と再生である。筋線維の壊死と再生に関しては以下のような説明がされている。ジストロフィン欠損に起因する膜の異常があり、細胞外液が細胞内に流入する。外液中には高濃度のカルシウムが存在するためそれが筋細胞に入ると筋肉は過収縮をおこす。これがopaque線維と考えられる。高濃度カルシウムが存在するとカルパインなどの酵素が活性化され自己消化を起こし、筋肉は崩壊し、貪食細胞の侵入を許すことになる。筋ジストロフィーでは筋再生が活発であるが、再生は壊死を代償しない。そのため筋線維は次第に数を減らし、末期には筋線維はほとんど消失し、脂肪組織と結合組織で置換される。骨格筋のみならず、心筋や横隔膜もおかされ、心不全または呼吸不全が死因のひとつとなる。
先天性筋ジストロフィー
出生時より筋力の低下を認めるものを先天性筋ジストロフィーと呼ぶ。
- 福山型 - 日本では先天性筋ジストロフィーの中で最も頻度が高い。多くは10歳代で死亡する。
- ウールリッヒ型
- メロシン欠損症
- インテグリン欠損症
- ウォーカーワールブルグ症候群
肢帯型筋ジストロフィー
- LGMD1A - 1D群
- LGMD2A - 2F群
三好型筋ジストロフィー (Miyoshi muscular dystrophy : MMD)
16 - 30歳ごろに発病し腓腹筋とヒラメ筋が侵される。初期症状は、つま先立ちができないジャンプすることができない、走ることが遅くなるなどの症状報告されている。発症後約10年で歩行が不可能となり、手の筋力も遠位から低下しやがて近位にも及んでくると言われているが、病状には違いがあり、発症後10年以上経過した方でも歩行可能の患者が報告されている。この病気は筋ジストロフィーの一種で血中のCK値が顕著に上昇する。原因遺伝子はdysferlinで常染色体劣性遺伝。肢帯型筋ジストロフィー2B型においてもdysferlinの異常が確認されている。肢帯型筋ジストロフィー2B型は、体幹に近い所から筋肉が萎縮するが、病状が進むにつれ三好型と同じように体幹から遠いい手や足にも筋肉の萎縮が現れる。現在、有効な治療法はないが、共に治療を受けることができる。このDysferlin異常で発症する病気をDysferlinopathyと呼ぶ。
顔面肩甲上腕型筋ジストロフィー
 | この節の加筆が望まれています。 |
常染色体優性遺伝。第4番染色体長腕に遺伝子座。原則として両親のどちらかが病気であるが、両親が全く正常で突然変異による発症と考えられる例が30%ある。病名のように顔面、肩甲部、肩、上腕を中心に障害される。進行すると腰や下肢の障害も生じ歩行困難となることもある。顔面筋の障害により閉眼力低下、口輪筋障害(口笛が吹けない)などを来たし、独特の顔貌(ミオパチー顔貌)を呈する。肩や上腕の筋萎縮が高度なのに比し前腕部は比較的保たれるため、ポパイの腕と形容される。下肢の障害は、下腿に強いもの、腰帯・大腿に強いものなどいろいろである。CK上昇は軽度である。比較的良性の経過をたどり、進行すると腰や下肢の障害も生じ歩行できなくなることもあるが、生命に関しては良好な経過をとる。筋症状以外では、感音性難聴、網膜血管異常の合併が高率であり、まれに精神遅滞やてんかんの合併がある。
筋緊張性ジストロフィー (myotonic dystrophy)
筋強直性ジストロフィーとも呼ばれる。常染色体優性遺伝を示す疾患で、マウスではmuscleblind-like(Mbnl)遺伝子の阻害により同様の症状が発現することが確認されている[1]。トリプレットリピート病の一種である。進行性に罹患筋の萎縮とミオトニアが見られる。有病率は10万人に1 - 5人、好発年齢は20 - 30歳代であるとされる。先天型では母からの遺伝による重症型がある。フロッピーインファントで発症。
- 症状
- 顔筋、舌筋、手内在筋のミオトニア(筋強直。筋の収縮が異常に長く続き、弛緩が起こりにくい現象のこと。手を強く握るとすぐには開けない、など。低温下で増強されるため、冷水中の雑巾絞り様動作が診断の一助になるという)や、咬筋・胸鎖乳突筋の筋萎縮(西洋斧顔貌)、側頭筋の筋萎縮(白鳥の頸)、または四肢遠位筋の筋萎縮を見る。ミオトニアは筋萎縮に先立って生じる。
- その他に、白内障などの眼症状、内分泌障害(耐糖異常、性腺萎縮(無精子症)、甲状腺機能低下)、精神薄弱、循環器障害、呼吸器障害、消化器障害、前頭部の脱毛など多彩な症状の見られる全身性疾患である。
- 検査
- 血清CK軽度上昇。筋電図にて筋原性変化を認め、また電極の刺入時に特徴的な筋強直性放電を認める(急降下爆撃音)。
- 治療
- 現在のところ、根本的治療法はない。対症的にプロカインアミド、フェニトイン、塩酸キニーネ、副腎皮質ステロイド剤などの投与を行う。
脚注
注釈
出典
- ^ トランスレーショナル・メディカルセンター (2013年5月9日). “国産初のアンチセンス核酸医薬品としてデュシェンヌ型筋ジストロフィー治療剤の臨床試験開始へ”. 2014年10月29日閲覧。
- ^ 国立研究開発法人 日本医療研究開発機構 (2020年3月27日). “デュシェンヌ型筋ジストロフィー治療薬(NS-065/NCNP-01、ビルトラルセン)の製造販売承認について”. 2020年7月10日閲覧。
参考文献
| 出典は列挙するだけでなく、脚注などを用いてどの記述の情報源であるかを明記してください。 |
- F.グレイ、U.デ・ジロラーミ、J.ポワリエ 編著 著、村山繁雄 監訳 編『エスクロール基本神経病理学』西村書店、2009年10月。ISBN 9784890133765。
- 埜中征哉『臨床のための筋病理』(第4版)日本医事新報社、2011年1月。ISBN 9784784950645。
関連項目
- World Community Grid - 治療支援のための分散コンピューティング
- レイバー・デイ・テレソン - 筋ジストロフィー患者の治療費捻出や社会参加を訴えるためのチャリティーテレビショーで、1966年からジェリー・ルイスを発起人として毎年開催されている
- 筋強直症候群
- 遠位型ミオパチー
- 国際宇宙ステーション - 筋ジストロフィー患者特有のタンパク質を分析、結晶化させるための宇宙実験が行われている。これによる新薬開発に期待が高まっている。
- 戸田達史 - 福山型先天性筋ジストロフィーの原因遺伝子やフクチンの研究で著名。
- 呉建 - 戦前に進行性筋ジストロフィー症の成因および治療について研究した。
- こんな夜更けにバナナかよ 筋ジス・鹿野靖明とボランティアたち
外部リンク
- 社団法人 日本筋ジストロフィー協会
- Remudy(Registry of Muscular Dystrophy) レムディ。臨床試験・治験を目的として、患者-製薬関連企業・研究者の橋渡しをする登録システム。
- 筋ジストロフィー(指定難病113) - 難病情報センター
- Atom's Home Page
| 国立図書館 | |
|---|---|
| その他 | |
 | この項目は、医学に関連した書きかけの項目です。この項目を加筆・訂正などしてくださる協力者を求めています(プロジェクト:医学/Portal:医学と医療)。 |

筋ジストロフィー
出典: フリー百科事典『ウィキペディア(Wikipedia)』 (2022/05/04 05:00 UTC 版)
筋病理では筋ジストロフィーにおいても反応性の細胞浸潤がしばしば認められる。特に顔面肩甲上腕型筋ジストロフィー(FSHD)、LMNA遺伝子変異による筋ジストロフィーやジスフェルリン遺伝子変異による筋ジストロフィーが有名である。LMNA遺伝子変異による筋ジストロフィーはエメリ・ドレフェス型筋ジストロフィーやLGMD1Bを呈する。またジスフェルリン遺伝子変異はLGMD2Bを呈する。原則としては炎症性筋疾患ではHLA-ABCが筋線維に発現するが筋ジストロフィーでは発現しないことが多い。
※この「筋ジストロフィー」の解説は、「筋炎」の解説の一部です。
「筋ジストロフィー」を含む「筋炎」の記事については、「筋炎」の概要を参照ください。
「筋ジストロフィー」の例文・使い方・用例・文例
筋ジストロフィーと同じ種類の言葉
- 筋ジストロフィーのページへのリンク


