炎症性筋疾患
(1)多発筋炎(polymyositis)、皮膚筋炎(dermatomyositis)
炎症性筋疾患(多発筋炎)は表4のように分類されています。
| I. 成人型多発筋炎 II. 成人型皮膚筋炎 III. 小児および若年型皮膚筋炎 IV. 膠原病を伴う皮膚筋炎 V. 膠原病を伴う多発筋炎 VI. 悪性腫瘍に伴う皮膚筋炎 VII. 悪性腫瘍に伴う多発筋炎 |
| (Banker と Engel,1986) |
| 表4 多発筋炎の分類 |
成人の多発筋炎(polymyositis)、皮膚筋炎(dermatomyositis)は臨床的に厳密な区別がつけがたいものが多いので、ここでは一括して説明します。
a.病因、病態、病理
原因不明の特発性のものと、結合織疾患や悪性腫瘍に伴うものがあります。罹患筋では筋線維の壊死・再生とともに、単核球の細胞浸潤を間質、血管周囲に認めます。また単核球は壊死線維の周囲に集積して存在することもあります。このような細胞はCD8陽性のT細胞(cytotoxic T cell)が多いことより、T細胞によって筋線維が直接傷害されると考えられています。
皮膚症状が特に顕著な筋炎は皮膚筋炎とよばれます。病理学的には血管炎が主で、しばしば虚血性の変化(筋束周辺萎縮、小梗塞像など)をみます(図29)。
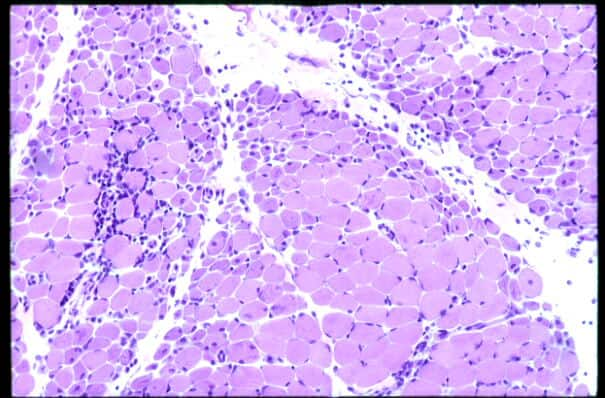 | 小さく黒く染まっているのは浸潤しているリンパ球。 筋束の周辺の筋線維が細くなっていることが(筋束周辺萎縮: perifascicular atrophy)診断的所見である。 |
| 図29:皮膚筋炎の病理 | |
主な臨床症状は筋力低下です。躯幹近位筋のことが多く、ごく例外的に筋力低下を局所的にみることがあります。頸部の屈筋群、咽頭筋がおかされることもまれでなく、その場合は嚥下困難をみます。急性期には発熱、筋痛、倦怠感、レイノー(Raynaud)現象を認めます。皮膚症状で典型的なものは上眼瞼に淡赤紫色の発疹(heliotrope rash)です。腱反射は消失ないし減弱します。
慢性に経過するものは、近位筋の筋力低下で気付かれます。筋ジストロフィーとの鑑別が困難なこともまれではありません。
成人例では約20%に腫瘍の合併があり、特に40歳以上で皮膚筋炎の場合はその可能性が高いといわれています。腫瘍が発見される以前に筋症状が出現することもあります。腫瘍の中では肺癌が特に多くみられます。結合織疾患としてはエリトマトーデス、慢性関節リューマチ、シェーグレン症候群が代表的です。
小児皮膚筋炎は成人の皮膚筋炎と異なり、悪性腫瘍を伴うことはなく、予後は良好です。皮膚症状は眼瞼周囲の紅斑、手足関節周囲の発疹です。症状は急性で、筋力低下は近位筋優位にみられます。病理学的には筋束周辺萎縮(perifascicular atrophy)と血管炎を主病変とします。ステロイドが著効しますので、早期診断、早期治療開始が重要です。
類肉腫性筋炎(granulomatous myositis)はサルコイドーシスとの関連性が深いと考えられています。サルコイドーシスは全身性の疾患で骨格筋の症状を伴うことはまれとされていました。しかし、症例によっては筋症状が前景に立つことが知られています。さらに筋内にサルコイド結節を証明しても全身性の所見に欠けることもあります。これらは類肉腫性筋炎として区別してよばれていますが、多分サルコイドーシスと同一なものではないかと考えられています。
c.検査所見
急性期には赤沈の亢進、白血球の増加があります。血清CK値は上昇します(皮膚筋炎では正常のこともある)。自己免疫疾患と合併した例では免疫グロブリン(α2、γなど)の増加があり、RA(リュウマチ)、LE因子が陽性となります。
d.治療
ステロイド剤が第一選択です。成人では60mg/日より開始し、症状、血清クレアチンキナーゼ(CK)値、赤沈値などの値をみて次の治療方針をたてます。激症で筋力低下が急速に進むものはステロイド大量点滴(パルス)療法、血漿交換が行われることもあります。少なくとも一ヶ月間継続します。次にステロイド抵抗例には免疫抑制剤を使用します。
慢性例では関節拘縮の防止、筋力低下防止のためのリハビリテーションが必要です。
(2)封入体筋炎(inclusion body myositis: IBM)
a. 病因、病態、病理
主として50歳以降の高齢者、特に男性に多くみられる特異な筋炎です。筋病理で、筋線維間へのリンパ球浸潤とともに、筋細胞の核内に細い管状(直径約20nm)の封入体をみることから、上記の名称が与えられました。封入体は電子顕微鏡でしか確認できません。電子顕微鏡でみると、核内だけでなく、細胞質にも封入体をみます。封入体をもっている筋線維は光学顕微鏡で見ると縁取り空胞(rimmed vacuole)(図37)を持っています。ですから筋生検で縁取り空胞をもつ筋線維の存在と筋炎の所見の両方を確認すれば診断が可能です。原因は慢性のウィルス感染説などがありますが、よく分かっていません。
b.臨床症状
多くは歩行の異常で気づかれます。大腿前面の筋力低下、筋萎縮、それに上肢では前腕部内側の筋萎縮、筋力低下がきます。ものが握りにくい、握る力が入らないことで気づかれることもあります。経過は人によって異なりますが、発症後数年で車いす生活となる人もいます。検査ではあまり特異的な変化はありません。血清クレアチンキナーゼ値も正常かやや上昇する程度です。
c.治療
副腎皮質ホルモンや免疫抑制剤など多くの試みがされていますが、あまり効果は期待できません。筋萎縮予防のリハビリが中心です。心臓や呼吸筋は侵されにくいので、生命的な予後はよいとされています。
筋炎
(炎症性筋疾患 から転送)
出典: フリー百科事典『ウィキペディア(Wikipedia)』 (2025/05/05 07:06 UTC 版)

| 筋炎 | |
|---|---|
| 概要 | |
| 診療科 | リウマチ学 |
| 分類および外部参照情報 | |
| ICD-10 | M60 |
| ICD-9-CM | 729.1 |
| OMIM | 160750 |
| DiseasesDB | 29473 |
| MedlinePlus | 001245 |
| MeSH | D009220 |
炎症性筋疾患または筋炎(myositis)は骨格筋に炎症性変化および障害をきたす疾患である。ウイルスや細菌などの感染が原因となる感染性筋炎(infectious myositis)と自己免疫が原因となる自己免疫性筋炎(autoimmune myositis)または特発性炎症性筋疾患(idiopathic inflammatory myopathy、IIM)、薬物や治療に関連する有害事象性筋炎に分類される。有害事象性筋炎は免疫チェックポイント阻害薬関連筋炎、スタチン関連免疫介在性壊死性ミオパチー、慢性移植片宿主病などがある。おもに自己免疫性筋炎に関して述べる。
歴史
1975年に発表されたBohanとPeter(ボアンとピーター)の診断基準に基づき、長らく皮疹のある皮膚筋炎(dermatomyositis)と皮疹のない多発筋炎(polymyositis)に分類されていた[1][2]。膠原病内科や皮膚科領域では多発(poly)と多発性(multi)の区別がなされていないため多発筋炎を多発性筋炎と記載されていることもある。OlsenとWartmannはさらに悪性腫瘍に伴う筋炎や封入体筋炎も自己免疫性筋炎に加えて分類した。その後、筋炎特異自己抗体(myositis-specific autoantibody、MSA)と他の膠原病でも見出される筋炎関連自己抗体(myositis-associated autoantibodies、MAA)に関する知見や筋病理所見の特徴が明らかになった。2003年にDalalasとHohfeldが炎症性筋疾患から封入体筋炎を除外したうえに皮疹と有無と筋病理を重視した多発筋炎と皮膚筋炎の診断基準を作成した[3]。2004年にヨーロッパ神経筋センター(European Neuromuscular Centre、ENMC)ワークショップでは皮膚筋炎、多発筋炎、封入体筋炎、免疫介在性壊死性ミオパチー、非特異的筋炎の5つに分類するENMC分類基準を作成した[4]。さらに2014年のヨーロッパ神経筋センターのワークショップでは抗合成酵素症候群(anti-synthetase syndrome、ASS)が独立したサブタイプとして追加された[5]。さらに2018年に皮膚筋炎の診断基準も改訂した[6]。ミクソウイルス抵抗性蛋白質A(myxovirus resistance protein A、MxA)はⅠ型インターフェロン(IFN-Ⅰ)で誘導される代表的な蛋白質である。骨格筋の筋線維におけるMxAの発現は筋束辺縁部萎縮(perifascicular atrophy)よりも皮膚筋炎の診断で感度・特異度ともにすぐれており2018年の改訂で診断基準にも含まれるようになった。皮膚筋炎は全身性エリテマトーデスや関節リウマチとともにⅠ型インターフェロノパチーとして認識されるようになった。全身性エリテマトーデスは病態形成にIFN-Ⅰが関与するうえ、C型肝炎の治療でIFN-α投与した際の副作用で全身性エリテマトーデス様の自己免疫現象が認められるためⅠ型インターフェロノパチーの代表疾患である。臨床的に多発筋炎と診断される例のほとんどが筋病理学的には免疫介在性壊死性ミオパチーであり、多発筋炎の組織学的定義は「CD8陽性T細胞の筋内鞘および非壊死性線維内部への浸潤を伴う」というものであるがこれを厳密に採用すると多発筋炎と病理診断される例はほとんどなくなった。従来、多発筋炎と病理診断されてきた例のほとんどは封入体筋炎であった[7]。そのため、自己免疫性筋炎は皮膚筋炎、抗合成酵素症候群、封入体筋炎、免疫介在性壊死性ミオパチーの4つのサブタイプに分類されるようになりつつある。自己免疫性筋炎は膠原病内科、皮膚科、小児科、脳神経内科と様々な診療科で診療される。2004年に上記4科の疫学者、生物統計学者が集まり、国際筋炎分類基準プロジェクト(International Myositis Classfication Criteria Project、IMCCP)を結成し、新たな国際筋炎分類基準を策定することになった。その結果はヨーロッパリウマチ学会、アメリカリウマチ学会によって2017年に承認された[8]。国際診断基準では無筋症性皮膚筋炎(amyopathic dermatomyositis、ADM)の定義が変更された点に注意が必要である。国際診断基準では筋力低下を認めず、皮膚筋炎に典型的皮疹をもつ症例はすべてADMとよぶ。
疫学
多発筋炎、皮膚筋炎の患者は日本では約17000人おり毎年1500人増加している。男女比は1対2.7と女性に多く中年期発症が多い。初期より間質性肺炎の合併が過半数で認められた。
病態
自己免疫性筋炎は種々の程度で筋炎、特徴的な皮膚症状、間質性肺炎を合併するスペクトラムである。がんの転移を説明する「種と土壌の理論(Seed and Soli model)」は多臓器症状を呈する炎症性筋疾患にも応用できる。細胞傷害性T細胞などの免疫細胞を種とすると、土壌としての条件が揃った特定の臓器へ集中して遊走し炎症を起こし、その全体像が自己免疫性疾患を成すという捉え方になる。
症状
全身症状
自己免疫性筋炎では全身症状として全身倦怠感や発熱、体重減少を呈しうる。体重減少は筋炎による筋量の低下や嚥下障害による摂食量低下も反映しうる。
骨格筋の筋力低下
亜急性もしくは慢性の経過で進行性の筋力低下を呈し、四肢近位筋、体幹筋、頸部屈筋、咽頭筋が優位に障害される。筋の自発痛や把握痛を認めることもある。具体的にはベッドからの起き上がり、しゃがみ立ち、階段昇降、上肢挙上(髪をとかす)などが困難になることで自覚されやすい。咽頭筋の障害は嚥下障害、発声障害をきたし誤嚥性肺炎を繰り返す例もある。頸部屈筋は自覚症状がなくとも筋力低下が鋭敏に検出されるため仰臥位で診察する。抗合成酵素症候群では関節痛を合併することも多い。
皮膚
皮膚の肉眼所見
皮膚筋炎の皮疹はケブネル現象の典型例と考えられている。ケブネル現象とは、正常皮膚へ掻爬・外傷・炎症・瘢痕化刺激に続き、その部位に原病と同類の皮膚症状が出現することをいう。ケブネル現象は皮膚筋炎のほか、尋常性乾癬、扁平苔癬、サルコイドーシス、モルフェアなどでも認められる。皮膚病変を起こす全身的素因に加えて、局所への機械刺激が加わると病変を形成すると解釈できる現象である。ゴットロン丘疹・ゴットロン徴候は関節伸側の機械的刺激を受けやすい部位に生じ、ヘリオトロープ疹は恒常的に機械運動が行われる眼瞼に生じる。メカニクスハンドも母指尺側と他4指の橈側に出るのが特徴的であり、物を掴む際に物と接触する面に生じるケブネル現象である。
- 手の皮膚所見

手の皮膚所見は最も診断的価値が高い。ゴットロン丘疹、ゴットロン徴候は関節背面に生じる皮疹で丘疹性変化を呈する場合にゴットロン丘疹、それ以外の場合(主に紅斑)にゴットロン徴候という。特に示指、中指のMP関節やPIP関節に好発する。爪郭部の所見、すなわち爪囲紅斑および爪上皮出血点も非常に重要である。爪囲の変化はゴットロン丘疹やゴットロン徴候に先行することが多い診断的価値は高い。爪郭部の所見は皮膚筋炎に特異的ではないが膠原病に特徴的である。掌側の指関節周囲に紫紅色の鉄棒豆様の皮疹が見られることがある。これを逆ゴットロン徴候といい、抗MDA5抗体陽性例に特徴的な皮疹である。メカニクスハンドは手指の側面、特に拇指尺側や示指橈側を中心に生じる手湿疹に類似した皮疹である。抗合成酵素症候群に特徴的とされる。
- 顔面・頭部の皮膚所見

ヘリオトロープ疹は上眼瞼に見られる浮腫性紅斑で色調は紫紅色から暗紅色を呈することが多いが浮腫のみのこともある。頬部、前額、耳介などに紅斑もしばしば認められる。
- 体幹の皮膚所見
体幹ではV徴候とよばれる前胸部、ショール徴候とよばれる肩から上背部の皮疹がよく知られている。
- 四肢の皮膚所見
四肢の関節背面(肘、膝)にも紅斑を生じこれらもゴットロン徴候と呼ばれる。
- 部位によらない皮疹
水疱、脂肪織炎、皮膚潰瘍、多型皮膚萎縮(ポイキロデルマ)、皮下石灰化沈着などが認められる。特に皮下石灰化沈着は抗NXP2抗体陽性例に多い。
皮膚病理所見
皮膚筋炎の皮膚病変の組織学的な変化は飛石状に断続的に認められる場合があるため、生検標本は比較的大きめにとり、有意な所見が認められない場合には複数の切片から標本を作成する。皮膚筋炎の皮疹の病理組織像として共通する基本的な所見は、表皮基底層の液状変性、真皮の血管周囲を中心とした炎症細胞浸潤、真皮のムチン沈着である。これらの所見は皮膚筋炎に特異的ではなく全身性エリテマトーデスとの皮膚病理所見で鑑別は困難である。
- 表皮基底層の液状変性
表皮基底層の液状変性とは基底層の角化細胞が空砲状に変性をきたした状態であり、皮膚筋炎の組織像の特徴的所見のひとつである。アポトーシスに陥った表皮細胞が好酸性のコロイド小体として認められることもある。このような変化は皮膚筋炎特異的ではなく、全身性エリテマトーデスや扁平苔癬でも認められる。
- 真皮炎症細胞浸潤
炎症細胞が真皮浅層の血管周を中心に散在性あるいは帯状に分布する。浸潤細胞の多くはCD4陽性T細胞であるがCD8陽性T細胞、マクロファージ、形質細胞様樹状細胞なども混じる。抗合成酵素症候群例では表皮内にCD8陽性T細胞が多く浸潤する。
- 真皮のムチン沈着
真皮にムチンが沈着するためHE染色で真皮の間質がやや好塩基性の紫がかった色を呈する。アルシアン青染色などでも確認できる。
肺・心臓・消化器の合併症
労作時の呼吸困難がある場合は間質性肺炎または心筋炎の合併を考慮する。間質性肺炎は報告にもよるが20~30%に合併すると言われる[9]。抗ARS抗体や抗MDA5抗体陽性例では間質性肺炎が必発と考えられている。抗合成酵素症候群は間質性肺炎が80%に認められるという報告もある。病理学的な区分では非特異的間質性肺炎(NSIP)が多い。
心筋炎の合併は心不全や不整脈などの症候例は約10%である[10]。重症の場合は致死的である。心電図や心筋逸脱酵素の測定や心臓超音波検査が必要となる。
消化器障害の合併は咽頭筋の障害が最も重要である。また炎症性腸疾患や特発性胆汁性肝硬変、特発性硬化性胆管炎を合併することがある。
内蔵悪性腫瘍
自己免疫性筋炎では内蔵悪性腫瘍の合併頻度が一般人口よりも5~7倍高い[11]。乳腺や下部消化管も含めたスクリーニングが必要である。
検査所見
血液検査
末梢血所見では軽度の白血球上昇や軽度の貧血を認めることがある。赤沈値やCRPなどの炎症反応は軽度の上昇にとどまることが多く、活動性指標としての意義は乏しい。むしろ炎症反応の上昇は感染症の合併を疑う。血清筋原性酵素(CK、LDH、AST、ALT、アルドラーゼ)の上昇は筋疾患を示唆する。特に感度が高く、疾患活動性ともっともよく相関するのがCKである。CKはアイソザイムに骨格筋由来のCM-MMと心筋由来のCK-MBがありCK-MB/CKが10%を超える場合は心筋障害の合併を疑う。腫瘍性疾患や慢性肝疾患で高CK血症をみとめることもある。これはIgG成分がCKに結合してCKのクリアランスが低下することで高CK血症をしめすものでマクロCKとよばれるが臨床的意義は不明である。血清フェリチンやKL-6は筋炎のマーカーではないが合併する間質性肺炎のマーカーである。
自己抗体
筋炎に特異的に検出される筋炎特異的自己抗体(MSA)と筋炎重複症候群に見いだされる筋炎関連自己抗体(MAA)が知られている。
| 筋炎特異的自己抗体 | 筋炎関連自己抗体 | |
|---|---|---|
| 皮膚筋炎 | TIF1-γ、MDA5、Mi-2、NXP-2、SAE | |
| 抗合成酵素症候群 | Jo-1、OJ、PL-7、EJ、KS、Ha、Zo、SC、JS | |
| 免疫介在性壊死性ミオパチー | SRP、HMGCR | ミトコンドリアM2 |
| 封入体筋炎 | cN1A | |
| オーバーラップ症候群 | Ku |
筋炎関連自己抗体(MAA)
筋炎関連抗体には抗核抗体、抵U1RNP抗体、抗SS-A/Ro抗体、抗Ku抗体、抗PM-Scl抗体、抗ミトコンドリアM2抗体、抗cN1A抗体などが知られている。欲に抗Ku抗体はオーバーラップ症候群、抗ミトコンドリアM2抗体は免疫介在性壊死性ミオパチー、抗cN1A抗体は封入体筋炎に関連する。
筋炎特異的自己抗体(MSA)
- 皮膚筋炎
皮膚筋炎の筋炎特異的自己抗体としては抗TIF1-γ抗体、抗MDA5抗体、抗Mi-2抗体、抗NXP-2抗体、抗SAE抗体が知られている。
- 抗合成酵素症候群
抗合成酵素症候群の筋炎特異的自己抗体としてはアミノアシルtRNA合成酵素(ARS)に対する自己抗体である抗ARS抗体が知られている。抗ARS抗体は総称であり理論上のものもふくめ10種類知られている。それは抗Jo-1抗体、抗OJ抗体、抗PL-7抗体、抗PL-12抗体、抗EJ抗体、抗KS抗体、抗Ha抗体、抗Zo抗体、抗SC抗体、抗JS抗体である。少数の例外を除き、抗合成酵素症候群では抗ARS抗体は1種類のみ検出され重複はしない。
- 免疫介在性壊死性ミオパチー
免疫介在性壊死性ミオパチーの筋炎特異的自己抗体としては抗SRP抗体や抗HMGCR抗体が知られている。
画像検査
- CT
全身の筋萎縮や間質性肺炎の有無を評価する。
- MRI
MRIは間質の線維化や炎症性変化による浮腫性変化などCTでは評価できない筋束内の構造の評価が可能である。筋炎にはかかせない検査である。T1WI、T2WI、STIRで撮影するのが通常である。筋束内で炎症を生じている線維分布と浮腫をSTIRで評価する。STIRで高信号であっても病理学的に炎症細胞浸潤があるとは限らない。Gd造影に関してはSTIRやT2WIに比べて浮腫性変化の検出率が高いとは言えず、サルコイドーシスや筋膜炎を想定していない場合、Gd造影は不要である。皮膚筋炎ではしばしば筋膜にアクセントのある浮腫性変化や皮下浮腫が認められることがある。
- シンチグラフィー
99mTc-MDPシンチグラフィー、あるいは骨シンチグラフィーは全身の骨格筋の炎症を一度に評価できる。
生理検査
- 針筋電図
針筋電図では安静時に線維自発電位や陽性鋭波や偽ミオトニー放電が認められる。これは分節性壊死や再生線維が関与する所見と考えられている。また随意収縮時に急速動員やBSAPパターンのMUPが認められる。
筋病理
筋生検はMRIで浮腫性変化がある部位から採取する[12]。高用量PSL投与前後の筋生検の病理像に関しての報告がある。免疫染色を行って両者を比較するとステロイド療法後に筋肉に浸潤しているリンパ球数が減り、筋肉内の炎症性サイトカインや血管内皮細胞での細胞接着因子の発現や筋肉のHLA-ABC発現の低下が示されている[13]。そのため筋生検は治療前に行うことが望ましい。筋病理に関して下記のようにまとめる。
筋原性変化
筋原性変化は炎症性筋疾患の他、ミオパチーなどでも認められる所見である。筋線維径のサイズは大小不同を呈し、筋線維の形は円形化する。慢性経過症例では100μm以上の肥大線維を認め、筋内鞘が開大し、間質の開大や線維化をみとめることもある。線維の中心部に核がある、いわゆる中心核を認めることも多い。壊死・再生筋をみとめる。NADH-TR染色では筋原線維間網の乱れが認められAcidP染色では再生線維で活性の上昇が認められる。
皮膚筋炎

筋原性変化が認められる。筋束辺縁部萎縮(perifascicular atrophy)が最もよく知られた診断的所見である。筋束辺縁部の筋線維の萎縮である。抗MDA5抗体陽性例では筋束辺縁部萎縮を認めないことが多い。筋束辺縁部萎縮周辺の筋線維はミトコンドリアやライソゾームが増加し細胞質が好塩基性に染色され、時にpunched-out vacuoleと呼ばれる空砲を有し、大型の内在核を伴っている。punched-out vacuoleは特に抗TIF-γ抗体陽性例で高頻度に認められる。筋束辺縁部の筋線維はミトコンドリアやライソゾームの増加を反映してNADH-TRで濃染する一方で、しばしばCOX活性が低下している。筋周鞘の血管周囲の単球浸潤もしばしば認められるが疾患特異性は低い。一部の症例では微小梗塞を認める。微小梗塞は小児例に多く、抗NXP-2抗体陽性例に多い。抗Mi-2抗体陽性例の筋病理は独自で、筋束辺縁部に壊死・再生筋が豊富に認められる。このような所見は筋束辺縁部壊死(perifascicular necrosis)と呼ばれる。また筋周鞘に浮腫が強い傾向があり、結合組織の断片化を認めるとともに、しばしばアルカリホスファターゼ活性が発現している。 免疫染色では筋細胞膜にHLA-ABCが発現するとともに内鞘毛細血管への膜侵襲複合体(MAC)沈着を認める。HLA-DRが一部の筋線維で発現した症例も存在するが稀である。正常ではHLA-ABCは血管内皮に発現するが筋細胞膜では発現していない[14]。全体の50%以上の筋線維の筋細胞膜にHLA-ABCの発現亢進を認める場合は検査陽性としたとき、筋炎の診断感度は100%であり、特異度は94%という報告もある[15]。HLA-ABCの筋線維の発現は、疾患活動初期より認め、炎症細胞浸潤に先立ち、疾患の慢性経過時にも残存することが知られている[15]。ミクソウイルス抵抗性蛋白質A(myxovirus resistance protein A、MxA)はⅠ型インターフェロン(IFN-Ⅰ)で誘導される代表的な蛋白質である。骨格筋の筋線維におけるMxAの発現は筋束辺縁部萎縮(perifascicular atrophy)よりも皮膚筋炎の診断で感度・特異度ともにすぐれており2018年の改訂で診断基準にも含まれるようになった[6]。皮膚筋炎は全身性エリテマトーデスや関節リウマチとともにⅠ型インターフェロノパチーとして認識されるようになった。 電子顕微鏡では血管内皮にtubuloreticular inclusions(TRIs)と呼ばれる管状構造物の集塊を認める[16]。
かつては血流障害の結果、筋束辺縁部萎縮が生じると考えられていたが反論が多い。Ⅰ型インターフェロンの下流遺伝子の発現亢進で筋束辺縁部萎縮が生じるという仮説もある。また皮膚筋炎で認められる炎症細胞はCD4陽性T細胞やB細胞が主体であり、CD8陽性T細胞を認めることは少ない。検出される自己抗体によって臨床症状や病理所見多少異なることが明らかになってきた。
| 自己抗体 | 臨床的特徴 | 病理学的特徴 |
|---|---|---|
| TIF1-γ | 成人で悪性腫瘍合併 | Perifascicular atrophy、毛細血管へのMAC沈着、punched-out vacuoles |
| MDA5 | 無筋症性皮膚筋炎 | Perifascicular atrophyは稀、毛細血管へのMAC沈着 |
| Mi-2 | 筋力低下、高CK血症 | Perifascicular necrosis、周鞘ALP発現、周鞘結合組織断片化、、毛細血管へのMAC沈着は稀 |
| NXP-2 | 若年性皮膚筋炎 | 微小梗塞 |
| SAE | 広範な紅斑 | ? |
抗合成酵素症候群
筋原性変化が認められる。筋束辺縁部に壊死・再生線維が分布する筋束辺縁部壊死(perifascicular necrosis)、筋周鞘の結合組織断片化、筋周鞘のアルカリホスファターゼ活性発現が特徴的である。抗Mi-2抗体陽性皮膚筋炎での所見と酷似するが免疫染色の所見は大きく異なる。抗合成酵素症候群では筋線維にMxAの発現が認められない。またHLA-ABCが比較的強く発現し、HLA-DRが発現する筋線維を認めることもある。抗合成酵素症候群ではⅠ型インターフェロン経路ではなくⅡ型インターフェロン経路の亢進が示唆されている[12]。
免疫介在性壊死性ミオパチー
筋原性変化が認められる。壊死・再生筋にマクロファージの浸潤が認められる。リンパ球浸潤は認めないか、あっても反応性の変化として説明が可能なものである。慢性に経過する例では間質の線維化や脂肪浸潤が認められる。免疫染色では筋線維膜でのHLA-ABCの発現増加が認められるが皮膚筋炎や封入体筋炎と比べると非常に軽度である。通常はHLA-DRの発現は認められない。一部の筋線維膜で膜侵襲複合体(MAC)沈着を認める。またp62が筋細胞質内で顆粒状に染まり、自己貪食に関わるシャペロン蛋白と共局在している。抗ミトコンドリアM2抗体陽性筋炎も病理学的には免疫介在性壊死性ミオパチーに分類せざるを得ない例が多い。
多発筋炎
その他の自己免疫性筋炎と同様に筋原性変化が認められる。特徴的であるのは筋内鞘主体に炎症細胞浸潤を認めるということである。筋束の外である筋周鞘に存在するリンパ球は非特異的であり診断的特異性は殆どない。HE染色では小型単核球が非壊死筋線維を取り囲み、内部に侵入する像を認める。 免疫染色ではCD8陽性T細胞が筋内鞘を主体としたスペースに浸潤し、HLA-ABCを発現している非壊死筋線維を取り囲み、筋線維内に侵入する像を認める。これをCD8/MHC class Ⅰ complexという。この所見は皮膚筋炎では認められず多発筋炎に特徴的な所見と考えられていた[4][17]。筋内鞘主体にCD68陽性マクロファージを認める。電子顕微鏡では非壊死筋線維に単核球が接し、同部位では筋線維の基底膜は消失している。つまり、単核球が筋線維の基底膜を破壊して、筋線維の細胞質に侵入していると考えられている。筋線維の筋原性変化、HLA-ABCの筋細胞膜への発現亢進所見のほか、CD8陽性T細胞が非壊死筋線維を取り囲み、筋線維内に侵入する像を認めることが特徴的かつ診断的と考えられている。ヨーロッパ神経筋センター(European Neuromuscular Centre、ENMC)の診断基準ではCD8陽性T細胞が非壊死筋線維を取り囲み、筋線維内に侵入する像を認めると確実な多発筋炎と診断される[4]。
多発筋炎の病態機序としてはCD8陽性T細胞が筋内鞘中心に侵入し、パーフォリンと呼ばれる物質を放出しながら筋線維の基底膜を破って線維の内部に入り込み、筋線維を障害すると考えられている[3]。皮膚筋炎と異なり多発筋炎では筋局所において細胞性免疫機序が存在する。臨床的な多発筋炎の多くは病理学的には壊死性ミオパチーである。「CD8陽性T細胞の筋内鞘および非壊死性線維内部への浸潤を伴う」という多発筋炎の組織学的な定義を用いると多発筋炎と病理学的に診断される例はほとんどなく、そのような所見を示す例の殆どが封入体筋炎である[7]。筋病理学的な立場では多発筋炎はもはや存在しない疾患という位置づけになっている[18][19][20]。
封入体筋炎
その他の自己免疫性筋炎と同様に筋原性変化が認められる。小型単核球が非壊死筋線維を取り囲み、内部に侵入する像を認める。本所見は後述する縁取り空砲と共に、封入体筋炎に診断的な所見の一つである。ゴモリ・トリクローム変法(modified Gomori trichrome、mGT)では赤色に染色される顆粒状物質で縁取られる縁取り空砲(rimmed vacuole)が認められる。縁取り空砲は変性した筋線維に存在し、封入体筋炎症例の全筋繊維の1~6%に認めると報告されている[21]。また、高頻度に赤色ぼろ線維(ragged red fiber、RRF)が認められる。赤色ぼろ線維はまだらに赤色に染色される筋線維であり、AcidP染色では空砲において高い活性を示す。 免疫染色を行うと、多発筋炎と同様にCD8抗体陽性T細胞が筋内鞘主体に浸潤し、HLA-ABCを発現している非壊死筋線維を取り囲み、筋線維内に侵入するCD8/MHC class Ⅰ complexが認められる。このことから封入体筋炎は局所的に細胞性免疫機序が存在することが示唆される。縁取り空砲の中や周囲の細胞質にコンゴーレッド染色で赤く染色されるβアミロイド(細胞内のアミロイド沈着)が認められる。βアミロイドの他にLC3やp62などのオートファジー関連蛋白質やTDP-43などの異常蓄積蛋白質の免疫染色で筋細胞質に顆粒状に認められる。筋線維内にアルツハイマー病様蛋白質や自己貪食や小胞体ストレスなどの要素が存在することは封入体筋炎において変性機序も存在することを示唆する[22]。 電子顕微鏡では筋細胞膜直下に空砲を認め、その内部にはグリコーゲン、膜様構造物、ミエロイド小体などが観察される。細胞質内または核内に直径15~20nmのfilamentous inclusionを認める。この封入体は封入体筋炎に特異的なものではなく、縁取り空砲をもつ細胞に高頻度に認められる。 2008年のMRC centre封入体筋炎ワークショップでは筋原性変化とHLA-ABCの筋細胞膜での発現亢進に加え非壊死筋線維への単核球の侵入像、縁取り空胞をもつ筋線維、細胞質内アミロイド沈着または電子顕微鏡でfilamentous inclusionを認めるものを病理所見から確実な封入体筋炎と診断される[23]。
LC3やp62などのオートファジー関連蛋白質やTDP-43などの異常蓄積蛋白質の免疫染色のほうがゴモリ・トリクローム変法の縁取り空胞や赤色ぼろ線維より感度がよい[24]。
診断
日本では厚生労働省の診断基準がよく用いられる。古典的にはBohen and Peterの診断基準[1]やTanimotoの診断基準[25]が知られている。2019年に小児・成人統一診断基準が作成された[26]。国際診断基準は従来の分類基準に比べて優れた感度と特異度を有している[8]。しかし各項目の点数を記憶するのが不可能であること、点数の総和から自己免疫性筋炎らしさを求めるのにグラフを参照する必要があるという欠点がある。そのためWeb計算機が作成され日本語版も公開されている。脳神経内科ではヨーロッパ神経筋センター(European Neuromuscular Centre、ENMC)の診断基準が重視される[4]
鑑別診断
筋ジストロフィー
筋病理では筋ジストロフィーにおいても反応性の細胞浸潤がしばしば認められる。特に顔面肩甲上腕型筋ジストロフィー(FSHD)、LMNA遺伝子変異による筋ジストロフィーやジスフェルリン遺伝子変異による筋ジストロフィーが有名である。LMNA遺伝子変異による筋ジストロフィーはエメリ・ドレフェス型筋ジストロフィーやLGMD1Bを呈する。またジスフェルリン遺伝子変異はLGMD2Bを呈する。原則としては炎症性筋疾患ではHLA-ABCが筋線維に発現するが筋ジストロフィーでは発現しないことが多い。
代謝性ミオパチー
糖原病、脂質代謝異常、ミトコンドリア病によるミオパチーも鑑別になる。
各論
皮膚筋炎(dermatomyositis)
皮膚筋炎は典型的には亜急性の経過でゴットロン徴候やヘリオトロープ疹といった特徴的な皮疹と近位筋優位の筋力低下を示す。5つの皮膚筋炎特異的自己抗体が同定されており、陽性自己抗体により特徴が多少異なる。成人例で最も多いのが抗TIF1-γ抗体であり高頻度に悪性腫瘍を合併する。小児では抗NXP-2抗体陽性が多い。筋症状に関しては通常は四肢近位筋や頸部の筋力低下を示す。無筋症性皮膚筋炎では筋症状が目立たず、その場合は抗MDA5抗体陽性であることが多い。CK値は様々であるが、抗MDA5抗体陽性例では正常値から軽度上昇であることが多い。抗Mi抗体陽性例では大半が1000以上である。骨格筋MRIでは、しばしば筋膜にアクセントを伴う浮腫性変化を認める。皮下浮腫を認める例もある。皮膚・筋以外の症状として重要なのは間質性肺炎である。特に抗MDA5抗体陽性の無筋症性皮膚筋炎では急速進行性間質性肺炎を合併することが多い。
若年性皮膚筋炎
成人の皮膚筋炎と異なり小児例は悪性腫瘍を合併しない、微小梗塞例の頻度が多い、皮下や筋膜の石灰化を伴うことがあるなど成人例と臨床的特徴が一部異なることから若年性皮膚筋炎と別疾患として扱われることがある。若年性皮膚筋炎の特徴とされてきた徴候のほとんどは抗NXP-2抗体陽性例の特徴であり、単に抗NXP-2抗体陽性例の頻度が高いことを反映しているだけかもしれない。
無筋症性皮膚筋炎
無筋症性皮膚筋炎は抗MDA5抗体陽性皮膚筋炎がよく知られているが抗ARS抗体陽性例や抗TIF1-γ抗体陽性例も含まれる。無筋症性皮膚筋炎に伴う間質性肺炎の予後は悪いと報告されているが抗MDA5抗体陽性例が多く含まれる影響と考えられる[27]。
抗MDA5抗体陽性皮膚筋炎
抗MDA5抗体陽性皮膚筋炎は潰瘍、脂肪織炎、関節炎、間質性肺炎を特徴とする皮膚筋炎のひとつの病型である。急速進行性間質性肺炎を合併することが多く予後が悪いことで知られている[28]。クラスター分析では重篤な急速進行性間質性肺炎を伴うタイプ、関節炎を伴うタイプ、血管障害を伴うタイプの3つのクラスターに分類されることが示された[29]。この疾患はIL-6、IL-10、IL-18、IFN-γ、TNF-αなどのサイトカインの上昇や画像所見がCOVID-19に類似しており[30]、MDA5がSARS-CoV-2の認識にも関連することから注目されている[31][32][33]。
抗合成酵素症候群(anti-synthetase syndrome、ASS)
抗アミノアシルtRNA合成酵素(aminoacyl-tRNA synthetase、ARS)抗体陽性の筋炎例を総称して抗合成酵素症候群(anti-synthetase syndrome、ASS)または抗ARS抗体症候群と呼ぶ。抗ARS抗体は理論上のものを含め10種類が知られている。抗Jo-1抗体、抗OJ抗体、抗PL-7抗体の頻度が高い。臨床的には、筋炎に加えて間質性肺炎、メカニクスハンドなどの皮膚症状、多発関節炎、発熱、レイノー現象などを合併する。抗合成酵素症候群の間質性肺炎は抗MDA5抗体陽性例で典型的に認められる急速進行性間質性肺炎と異なり慢性に経過することが多い。皮膚筋炎では多発関節炎やレイノー現象を認めることは稀であり鑑別に有用である。筋症状は抗Jo-1抗体、抗OJ抗体、抗PL-7抗体で目立つ傾向があり、CKも高値になる。抗EJ抗体や抗PL-12抗体陽性例では間質性肺炎主体で筋炎症状は軽度であることが多い。CKも正常か、上昇しても軽度である。骨格筋MRIでは皮膚筋炎と同様、浮腫性変化は筋膜主体であることが多い。皮下にも浮腫を認めることがある。
免疫介在性壊死性ミオパチー(immune-mediated necrotizing myopathy、IMNM)
免疫介在性壊死性ミオパチーに特異的な自己抗体としては抗SRP抗体と抗HMGCR抗体が知られている。歴史的には抗HMGCR抗体はスタチン誘発性免疫介在性壊死性ミオパチーに特異的な抗体として報告されたが、その後検討では抗HMGCR抗体陽性の免疫介在性壊死性ミオパチーのうちスタチン内服歴があったのは18%に過ぎなかった。スタチン内服は免疫介在性壊死性ミオパチーのリスク因子ではあるが原因とは言えない。SRPもHMGCRも筋線維膜上に発現しており、自己抗体がこの抗原に結合する[34]。その結果C1qが誘導され、古典経路を介して順次補体が活性化される。最終的にC5b-9からなる膜侵襲複合体(membrane attack complex、MAC)が筋線維膜上に形成され、筋線維膜に穴があくため筋線維が壊死する[35]。
抗SRP抗体陽性壊死性ミオパチー、抗HMGCR抗体陽性壊死性ミオパチーのいずれも30歳代およびそれ以上の例が約90%を占める。小児例は筋ジストロフィーとの鑑別が非常に難しく筋生検を含む検査が必要となる。 典型的には亜急性に近位筋優位に筋力低下をきたす。筋症状としては筋力低下のほか、筋萎縮を認めることが特徴的で、比較的筋痛も伴うことが多い[36][37]。傍脊柱筋、嚥下に関係する筋や顔面の障害も伴うことがある。また首下がりや嚥下障害もよく認められる[38][39]。慢性に経過した一部の抗SRP抗体陽性例は顔面肩甲上腕型筋ジストロフィーが鑑別となる。皮疹、関節炎、間質性肺炎、心筋炎を伴うこともあるが筋外症状は比較的少ない。CKは大半の症例で1000以上の高値を示す。骨格筋ではびまん性の淡い浮腫性変化を筋内に認めることが多い。
抗ミトコンドリアM2抗体陽性筋炎
抗ミトコンドリアM2抗体は原発性胆汁性肝硬変に特徴的な自己抗体である。慢性経過で心筋障害、呼吸筋障害、骨格筋萎縮が目立つなど特徴のある筋炎として知られている[40]。頸部・体幹の筋力低下が目立ち、しばしば首下がりや呼吸不全を呈するため運動ニューロン病やネマリンミオパチーが鑑別になる。病理学的には免疫介在性壊死性ミオパチーに類似するが肉芽腫を高率に認めたという報告もある[41]。
免疫チェックポイント阻害薬関連筋炎
免疫チェックポイント阻害薬の免疫関連有害事象(immune-related adverse events、irAE)として免疫チェックポイント阻害薬関連筋炎が知られている。眼瞼下垂や複視が多く初発症状が重症筋無力症に似るのが特徴である。病理学的には免疫介在性壊死性ミオパチーに類似するが壊死・再生線維が数本単位でまとまって存在する傾向を認める[42]。
封入体筋炎(inclusion body myositis、IBM)
50歳以降に発症し、男性にやや多い。手指屈筋群、特に深指屈筋が障害されやすくボタンをかけられない、ペットボトルのキャップを開けられないといった訴えが多い。大腿四頭筋も障害されることが多い。進行性の筋力低下と筋委縮を認め、しばしば症状は左右非対称である。発症5年で日常生活に支障をきたす。嚥下障害は60%以上に認められるが呼吸筋や心筋は障害されにくい。血清CK値は正常から正常上限10倍程度まで増加する。筋MRIでは大腿四頭筋および腓腹筋内側頭に脂肪置換と浮腫性変化を認める。大腿四頭筋のうち大腿直筋は相対的にやや保たれる傾向がある。細胞質5’-ヌクレオチダーゼ(cN1A)に対する自己抗体が一部で検出される。病態機序は不明であるが変性や蛋白分解経路の異常、免疫系の異常が示唆されている[43]。
オーバーラップ症候群(overlap syndrome)
膠原病に筋炎を合併することがある。2つ以上の膠原病の診断基準を満たした場合はオーバーラップ症候群という。合併する筋炎は免疫介在性壊死性ミオパチー、皮膚筋炎、非特異的筋炎など様々であるが免疫介在性壊死性ミオパチーの頻度が高い。このような例のなかには抗Ku抗体陽性例が一定数存在する。
肉芽腫性ミオパチー(granulomatous myopathy)
肉芽腫病変は様々な筋疾患で認められる[44][45]。特発性、サルコイドーシス、感染症、悪性腫瘍、膠原病、炎症性腸疾患、原発性胆汁性胆管炎、クリオフィブリノーゲン症、異物などで起こり得る。また封入体筋炎や抗ミトコンドリアM2抗体陽性筋炎など炎症性筋疾患でもみとめられる。肉芽腫性筋ミオパチーを起こす悪性腫瘍としては悪性リンパ腫、膠原病としては多発血管炎性肉芽腫症、強皮症、関節リウマチ、炎症性腸疾患としてはクローン病があげられる。また肉芽腫性ミオパチーを起こす感染症は多く、粟粒結核、第3期梅毒、トキソプラズマ症、ニューモシスチス・イロベチイ、ブルセラ症、HTLV-1、クリプトコッカス症などがあげられる。乾酪性の肉芽腫を認めた場合は結核など抗酸菌による肉芽腫を考慮するが日本では筋内に認める肉芽腫をほぼ非乾酪性肉芽腫である。筋内に非乾酪性肉芽腫を認めた場合はまずはサルコイドーシスを疑う。筋内の非乾酪性肉芽腫はサルコイドーシスだけではなく、封入体筋炎、抗ミトコンドリアM2抗体陽性筋炎などでも認められる。筋のみで非乾酪性肉芽腫が認められる場合はサルコイドーシスと診断するのは難しい。
GVHD筋炎
造血幹細胞移植後の合併症としてGVHD筋炎が知られている[46][47]。他の炎症性筋疾患とは異なる病理所見を示す[48]。
治療
自己免疫性筋炎の治療としてはPSLを第一選択とすることは世界中でコンセンサスが得られている。Dalakasが2010年にレビューで提唱した治療指針がよく知られている[49]。この治療指針ではPSLを1mg/kgの高用量で開始し、重症例ではステロイドパルス療法で開始する。初期用量を3~4週継続し、2~3週おきに5~10mgのペースで減量すると記載されている。ステロイド抵抗例やPSL減量中に悪化があった場合は免疫グロブリン大量療法や免疫抑制剤の併用やリツキシマブを検討する。もうひとつよく知られた治療指針がメイヨークリニックの治療プロトコールである[50]。免疫抑制剤を初期から併用することとPSLの減量速度が比較的早いことが特徴である。免疫介在性壊死性ミオパチーに関しては2016年のENMCワークショップで治療アルゴリズムが提唱された[51]。抗SRP抗体陽性免疫介在性壊死性ミオパチーのリツキシマブ投与の効果はばらつきがあり、数回の投与で寛解を得る場合もあるがリツキシマブ定期投与が必要であった例まである[52]。抗SRP抗体陽性免疫介在性壊死性ミオパチーには77.8%で有効であったが抗HMGCR抗体陽性免疫介在性壊死性ミオパチーでは43.8%でのみ有効であった[53]。免疫介在性壊死性筋症のうち23.5%でリツキシマブ投与後に感染症が認められ5.9%が死亡している。免疫疾患を対象とした大規模検討[54]ではリツキシマブ投与後に17.3%に感染症が認められ、4.1%が死亡している。免疫介在性壊死性ミオパチーに対するリツキシマブの安全性はその他の免疫疾患とほぼ同等と考えられる。抗HMGCR抗体陽性免疫介在性壊死性ミオパチーでは免疫グロブリン療法の長期的な反復投与がしばしば行われている[51]。2年間の治療を行っても半数以上の免疫介在性壊死性ミオパチーの症例では不十分な神経学的改善にとどまっているという報告がある[38]。
参考文献
- 臨床のための筋病理 第5版 ISBN 9784784950669
- 多発性筋炎・皮膚筋炎 最新医学社 診断と治療のABC 81巻 別冊号
- 多発性筋炎・皮膚筋炎診療ガイドライン(2020 年暫定版)
- 免疫関連有害事象irAEマネジメント ISBN 9784765318648
- Curr Opin Neurol. 2020 Oct;33(5):590-603. PMID 32852298
- Curr Opin Neurol. 2019 Oct;32(5):704-714. PMID 31369423
脚注
- ^ a b N Engl J Med. 1975 Feb 13;292(7):344-7. PMID 1090839
- ^ N Engl J Med. 1975 Feb 20;292(8):403-7. PMID 1089199
- ^ a b Lancet. 2003 Sep 20;362(9388):971-82. PMID 14511932
- ^ a b c d Neuromuscul Disord. 2004 May;14(5):337-45. PMID 15099594
- ^ Neuromuscul Disord. 2015 Mar;25(3):268-72. PMID 25572016
- ^ a b Neuromuscul Disord. 2020 Jan;30(1):70-92. PMID 31791867
- ^ a b Neurology. 2003 Aug 12;61(3):316-21. PMID 12913190
- ^ a b Ann Rheum Dis. 2017 Dec;76(12):1955-1964. PMID 29079590
- ^ Am J Respir Crit Care Med. 2001 Oct 1;164(7):1182-5. PMID 11673206
- ^ Int J Cardiol. 2011 May 5;148(3):261-70. PMID 20826015
- ^ N Engl J Med. 1992 Feb 6;326(6):363-7. PMID 1729618
- ^ a b Curr Opin Neurol. 2019 Oct;32(5):704-714. PMID 31369423
- ^ Arthritis Rheum. 2000 Feb;43(2):336-48. PMID 10693873
- ^ Lancet. 1985 Feb 16;1(8425):361-3. PMID 2857418
- ^ a b J Clin Pathol. 2012 Jan;65(1):14-9. PMID 22075187
- ^ J Neurol Sci. 1974 Nov;23(3):391-402. PMID 4427123
- ^ Arthritis Res Ther. 2010;12 Suppl 1(Suppl 1):S4. PMID 20392291
- ^ JAMA Neurol. 2018 Dec 1;75(12):1528-1537. PMID 30208379
- ^ Curr Opin Neurol. 2019 Oct;32(5):704-714. PMID 31369423
- ^ Curr Opin Neurol. 2020 Oct;33(5):590-603. PMID 32852298
- ^ Muscle Nerve. 2006 Oct;34(4):406-16. PMID 16823856
- ^ Autoimmunity. 2008 Dec;41(8):563-9. PMID 18958757
- ^ Neuromuscul Disord. 2010 Feb;20(2):142-7. PMID 20074951
- ^ Acta Neuropathol Commun. 2013 Jul 1;1:29. PMID 24252466
- ^ J Rheumatol. 1995 Apr;22(4):668-74. PMID 7791161
- ^ 多発性筋炎・皮膚筋炎診療ガイドライン(2020 年暫定版)
- ^ Curr Rheumatol Rep. 2014 Dec;16(12):465. PMID 25366932
- ^ J Am Acad Dermatol. 2018 Apr;78(4):776-785. PMID 29229575
- ^ Neurology. 2020 Jul 7;95(1):e70-e78. PMID 32487712
- ^ Eur Respir J. 2020 Sep 24;56(3):2001618. PMID 32631836
- ^ Trends Microbiol. 2019 Jan;27(1):75-85. PMID 30201512
- ^ Front Immunol. 2020 May 8;11:939. PMID 32574256
- ^ J Virol. 2020 Jun 16;94(13):e00099-20. PMID 32295922
- ^ Neurology. 2018 Feb 6;90(6):e507-e517. PMID 29330311
- ^ Ann Rheum Dis. 2019 Jan;78(1):131-139. PMID 30309969
- ^ Ann Rheum Dis. 2006 Dec;65(12):1635-8. PMID 16679430
- ^ Arthritis Rheum. 2004 Jan;50(1):209-15. PMID 14730618
- ^ a b Semin Arthritis Rheum. 2019 Dec;49(3):420-429. PMID 31109639
- ^ Mayo Clin Proc. 2017 May;92(5):826-837. PMID 28473041
- ^ Brain. 2012 Jun;135(Pt 6):1767-77. PMID 22561642
- ^ Ann Neurol. 2017 Apr;81(4):538-548. PMID 28224701
- ^ J Autoimmun. 2019 Jun;100:105-113. PMID 30862448
- ^ 封入体筋炎 診療の手引き
- ^ Am J Clin Pathol. 1999 Jul;112(1):63-8. PMID 10396287
- ^ Autoimmun Rev. Apr-May 2014;13(4-5):372-4. PMID 24424169
- ^ Biol Blood Marrow Transplant. 2015 Mar;21(3):389-401.e1. PMID 25529383
- ^ Bone Marrow Transplant. 2009 Jan;43(2):159-67. PMID 18762758
- ^ Muscle Nerve. 2021 Jun;63(6):852-860. PMID 33651380
- ^ Nat Rev Rheumatol. 2010 Mar;6(3):129-37. PMID 20125096
- ^ Mayo Clin Proc. 2013 Jan;88(1):83-105. PMID 23274022
- ^ a b Neuromuscul Disord. 2018 Jan;28(1):87-99. PMID 29221629
- ^ Arthritis Care Res (Hoboken). 2010 Sep;62(9):1328-34. PMID 20506493
- ^ Ther Adv Neurol Disord. 2021 Mar 12;14:1756286421998918. PMID 33786066
- ^ Clin Infect Dis. 2021 Mar 1;72(5):727-737. PMID 32067031
関連項目
- ミオパチー (筋疾患)
- 筋病理学
- 免疫介在性壊死性筋症
外部リンク
炎症性筋疾患と同じ種類の言葉
- 炎症性筋疾患のページへのリンク
