先天性ミオパチー
名のごとく乳幼児期早期からの筋力、筋緊張低下があり、多くは歩行を獲得しますが、以後も筋力低下が持続する疾患です。本症は進行性がなく、予後良好であると考えられ、以前の教科書には先天性非進行性ミオパチーと「非進行性」の言葉がつけられていました。しかし、多くの患者さんを長期にフォローすると緩徐ですが進行性のものが多く、また呼吸不全に陥り易いなどから「非進行性」の言葉が今は取り除かれています。
a.病因、病態、病理
本症は病理学的特徴からいくつかの型に分けられています(表3)。
| ネマリン ミオパチー | セントラル コア病 | ミオチュブラー ミオパチー | CFTD *1 | その他の ミオパチー *2 | 筋肉特異構造 を示さないもの *3 | |
| 遺 伝 常染色体優性 常染色体劣性 X連鎖劣性 | + + | + + | + + + | + + | + | + + |
| 発育・発達の遅れ | + | + | + | + | + | + |
| 筋力低下 外眼筋 顔面筋 咽頭・舌・頸筋 近位筋優位 | + + + | + + + | + + + + | + + + | + + + | + + + |
| 筋 病 理 筋線維の大小不同 タイプ1線維優位 小径タイプ1線維 | + + | + + + | + + + | + + + | + + + | + − +*4 |
| 重症例の存在 | + | − | + | + | + | |
| *1先天性筋線維タイプ不均等症,*2 Fingerprint,multicore,reducing body,sarcotubular
myopathy など, *3 minimal change myopathy ともいわれる,*4 タイプ1,2線維とも小径,筋原線維の配列の乱れ | ||||||
| 表3:先天性ミオパチーの臨床と病理 | ||||||
 | 正常筋(左)とネマリンミオパチー(右)の生検筋のGomoriトリクローム変法染色(筋疾患の診断には最も大切な染色。 簡単で30分以内に結果が出る)。 本症では筋線維内に赤黒色に染まる糸状ないし顆粒状の物質を多く認め、それが診断的意義をもつ。 |
| 図26:ネマリンミオパチーの病理 | |
☆セントラルコア病(central core disease)は主に常染色体優性遺伝をとると考えられています。その遺伝子座は第19染色体にあって、リアノヂン(ryanodine)受容体遺伝子に変異がみられています。筋線維の中心部に筋小胞体やミトコンドリアがなく酸化酵素染色(NADH-TRなど)で中央部が果物の芯(core)をみるように染色されないのが特徴とされています(図27)。
 | 正常の骨格筋をNADH-TR染色すると濃く染まるタイプ1線維とタイプ2線維がモザイクをなしてみられる(図16左参照)。 セントラルコア病ではほとんど全ての線維が、濃染するタイプ1線維で、筋線維の中心に酵素活性が無く果物の芯(コア)のように見える。 |
| 図27:セントラルコア病の病理 | |
☆先天性筋線維タイプ不均等症(congenital fiber type disproportion)は上記のような筋線維内の異常な封入体や構造異常がなく、タイプ1(赤筋)線維がタイプ2(白筋)線維より12%以上の差をもって小径である場合の診断名です。もちろん患者さんは乳児期から筋緊張低下、筋力低下があり、発達が遅れています。
上記何れの疾患でも共通な重要な病理学的所見はタイプ1線維がタイプ2線維より小径で、タイプ1線維の数が正常上限の55%以上を占める(タイプ1線維優位:type 1 fiber predominance)ことです。さらに、筋線維径は全体に細く、未熟で未分化なものが多く存在します。筋線維タイプ分布の異常と未熟性が筋力、筋緊張低下の原因と考えられています。
b.臨床症状
先天性ミオパチーはどの病気でも、乳児重症型、良性先天型、成人発症型の3型に分けられています。上記疾患(ネマリンミオパチーとかセントラルコア病とか)は病理学的には互いに明らかに所見が異なり、また遺伝子座も異なっているのに、臨床症状はきわめてよく似ています。たとえばネマリンミオパチーの患者さんと先天性筋線維タイプ不均等症の患者さんをみても、よく似ていて臨床的には鑑別が困難です。
1)乳児重症型(severe infantile form)
最もよく知られているのはネマリンミオパチーとミオチュブラーミオパチーです。ネマリンミオパチーでは多くは、常染色体劣性遺伝をとると考えられています。重症型の10%位にアクチン遺伝子の変異があるようです。前に述べたように、ミオチュブラーミオパチーの場合はX連鎖劣性遺伝をとるので、ほとんどが男児です。
新生児期からの呼吸困難、哺乳力低下があり、人工換気、経管栄養を必要とします。細長く、表情のない顔をしていて、高口蓋を認めます。手足の動きはほとんどみられません。手、足関節の拘縮、先天性股関節脱臼をしばしばみとめ、腱反射は消失しています。予後は不良で、大多数は1歳までに死の転帰をとります。
2)良性先天型(benign congenital form)
先天性ミオパチーの大半の患者さんはこの良性先天型に属します。乳児期に発達の遅れがあり、筋力・筋緊張が低下したいわゆるフロッピーインファント(floppy infant)のことが多いです。歩行開始も遅れ、1歳半を過ぎることが多いです。歩行開始後も走れない、階段の昇降に手すりがいるなど筋力低下は持続します。なかには症状がほとんどなく、よほど専門的な知識を持つ医師でしか診断できないような軽症例もあります。筋力低下は非進行性か、あるいは進行してもとても緩徐です。
筋力低下は全身にありますが、頸部屈筋が弱い(寝ていて頭がもちあがらない)のが特徴的です(図28)。顔面罹患がありますので、細長い顔で表情に乏しく、高口蓋があります。ただし、セントラルコア病では顔面筋罹患は少なく、正常と変わらないことが多いです。
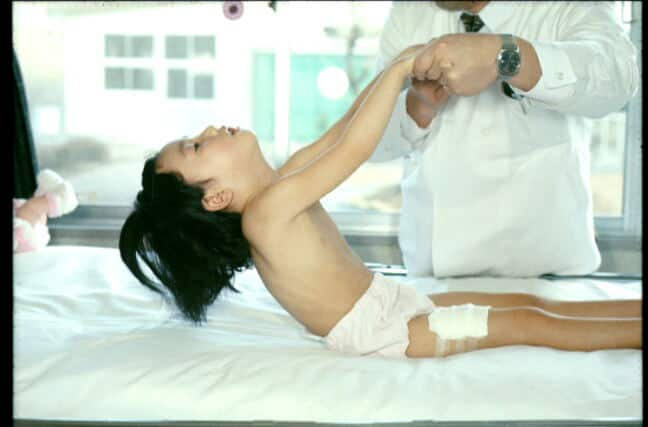 | 歩行は可能であるが、走れない、階段昇降困難を認めて診断のため来院した。 呼吸筋が弱く、さらに寝た位置から引き起こしても頭がついてこない(頸部屈筋の筋力低下)があるので先天性ミオパチーを疑われ筋生検し、診断が確定された。 |
| 図28:ネマリンミオパチー良性先天型 | |
関節拘縮はしばしば病初期から認めます。セントラルコア病では四肢筋の筋力低下は軽度なのに、強い脊柱変形(側彎)をみることがあります。
検査所見では、疾患特異的異常はありません。血清クレアチンキナーゼ(CK)値は正常ないし軽度上昇、筋電図は正常ないし筋原性所見を示します。
3)成人型(adult onset form)
ネマリンミオパチー、ミオチュブラーミオパチー(中心核病)での報告があります。良性先天型で幼少期にはきわめて症状が乏しく、成人になって急性増悪したものと、成人発症の2型があると考えられています。成人発症のものは病理所見だけで診断されていますので、良性先天型のような遺伝性はないのでしょう。原因も炎症、中毒などが考えられています。
先天性ミオパチー
出典: フリー百科事典『ウィキペディア(Wikipedia)』 (2024/12/30 07:01 UTC 版)
先天性ミオパチー (せんてんせいミオパチー、英: congenital myopathy) は、フロッピーインファント(ヒポトニア)を起こす疾患の一種として知られる疾患群である。先天性ミオパチーは非進行性で、筋病理で特徴ある所見を得る。
フロッピーインファントの分類
フロッピーインファントは筋力低下があるもの (paralytic) と筋力低下を伴わないもの (nonparalytic) に分類される。筋力低下を伴わないものは、脳性麻痺、精神遅滞、異染性白質ジストロフィー、セロイドリポフスチン症などの中枢神経変性疾患と、ダウン症候群、プラダー・ウィリー症候群などの染色体異常があり、中枢神経系の異常が原因疾患である場合が多い。筋力低下を伴うものは神経原性と筋原性に分類される。神経原性は、ウェルドニッヒ・ホフマン病、先天性髄鞘形成不全ニューロパチーなどがある。筋原性は広義の先天性ミオパチーと称され、糖原病、ミトコンドリア病、先天性筋ジストロフィー(特に福山型が有名)と、狭義の先天性ミオパチーに分類される。
狭義のミオパチーは病理学的な特徴により、ネマリンミオパチー、セントラルコア病、重症乳児型ミオチュブラーミオパチー、中心核病、先天性筋線維タイプ不均等症 (CFTD)、その他のミオパチー (fingerprint, multicore, reducing body, sarcotubular myopathy)、筋内特異構造を示さない minimal change myopthy、などに細分される。
先天性ミオパチーの共通点
多くの先天性ミオパチーは臨床像、病理像に共通点がある。臨床像の病型は3分類される。
- 重症乳児型 (severe infantile form)
新生児期から呼吸障害、嚥下障害を認め、多くは1歳以下で死亡するタイプである。セントラルコア病では稀である。
- 良性先天型 (moderate cogenital)
乳児期早期から筋緊張と筋力低下があり、発育、発達の遅れなどから異常を発見する。処女歩行も遅れ、歩行開始後も走れない、転びやすい、階段昇降困難など歩行の異常が持続する。顔面筋罹患があると表情は乏しくなり、外眼筋が侵されると眼球運動障害を認める。ほとんど全ての症例で高口蓋を認め、関節拘縮、脊柱管異常も合併しやすい。筋力低下が遠位優位な場合もある。先天性ミオパチーの大半はこのタイプで、非進行性ないし緩徐な進行を示す。
- 成人発症型 (adult onset)
病因は、良性先天型が極めて緩徐であった場合と、成人になって急速に進行して筋力低下が顕著となる場合もある。
筋病理では疾患分類が可能な特徴的所見もあるが、先天性ミオパチーの共通所見も知られる。共通所見はタイプ1線維優位で、タイプ1線維(赤筋)がタイプ2線維(白筋)よりも小径である。
先天性ミオパチーの各論
- ネマリンミオパチー
gomoriトリクローム変法で紫から黒赤色のネマリン小体が認められる特徴があるが、ネマリン小体は特異的所見ではない。HE染色はネマリン小体の見逃しが多い。アクチンフィラメントに関連する蛋白の遺伝子変異の所見が多い。
- セントラルコア病
常染色体優性遺伝で、リアノジン受容体遺伝子の変異を90パーセント (%) 以上で認める。乳幼児で死亡する重症例の報告はなく比較的軽症で、高口蓋を認めない症例もある。悪性高熱症で常染色体優性遺伝を示すもので、本症と同様の遺伝子変異を認める。
- 中心核病
顔面筋罹患が多く、約30%の症例で眼球運動障害や眼瞼下垂を認める。他の先天性ミオパチーに比して、中枢神経症状の合併が多く、10%で精神遅滞が認め、てんかんを伴う症例報告がある。
関連項目
参考文献
- エスクロール基本神経病理学 ISBN 9784890133765
- 臨床のための筋病理 第4版 ISBN 9784784950645

先天性ミオパチーと同じ種類の言葉
- 先天性ミオパチーのページへのリンク
