肢帯型筋ジストロフィー
デュシェンヌ型、ベッカー型、顔面肩甲上腕型、先天型筋ジストロフィーでは患者さんを診察すれば特徴的な臨床像があるので、臨床症状や筋生検の所見から診断できます。たとえばデュシェンヌ型では、患者は男児で3歳位から症状があり、ふくらはぎに筋肥大がある。筋生検すれば筋ジストロフィーの所見があり、ジストロフィンが欠損しているなどからです。しかし、そのような特徴的な所見がなく、ただ近位筋が好んで侵される筋ジストロフィーの患者さんが沢山おられます。診察してみて、今まで知られているどのタイプの筋ジストロフィーとも診断できない時、私たちは肢帯型筋ジストロフィーとよんでいます。多くは常染色体劣性遺伝ですが、まれには優性遺伝もあります(国立精神・神経センターのDNA診断・治療室の統計では優性遺伝は5%以下です)。このような本症の位置づけの経過からも解るように、肢帯型の原因は沢山ある(多因性である)と以前から考えられていました。近年の分子生物学的な進歩は見事にそれを証明したのです。表2には遺伝子の変異から分類されてきた肢帯型筋ジストロフィーの一覧表です。
これからも次々と肢帯型の患者さんの中から新しい遺伝子の異常がみつかり、肢帯型筋ジストロフィーの種類はもっと増えるでしょう。表では優性遺伝は(5%以下とすくないのですが)LGMD1、劣性遺伝をとるものはLGMD2に分類されています。肢帯型といっても、その原因は多岐にわたることが明らかにされたのです。遺伝子診断や筋生検の免疫組織化学的染色により、肢帯型の患者さんの70-80%は近い将来、あの複雑な分類のどのタイプに当てはまるか決定できるようになると言われています。
a.病因、病態、病理
上で述べたように肢帯型は原因が多岐にわたっているので、筋生検で免疫組織学的染色をしたり、遺伝子解析をしても半数以上の人は原因が何か分かりません。優性遺伝をとるものには5つの型が知られています。日本ではこの5つの型に相当する病気を持った人は少数しか報告されていません。 常染色体劣性遺伝をとるLGMD2Aは蛋白分解酵素であるカルパイン3遺伝子に変異がみられています。カルパイン3という酵素は筋肉が収縮するときに必要な細い糸のような構造物の一つであるコネクチン(タイチンともいいます)を調節しています。しかし、この病気ではコネクチンは正常に発現されています。この酵素が異常となると、なぜ筋線維が壊死なるのかそれは不明です。いろいろ調べてみると肢帯型の患者さんの30%位はこのカルパイン3の遺伝子変異をもっていることが明らかにされています。カルパイン3遺伝子に変異をもった肢帯型のことはカルパイノパチーともよばれます。 LGMD2Bはジスフェルリン(dysferlin)という遺伝子に変異があり、ジスフェルリンが欠損する病気です。このジスフェリルンは筋細胞膜にあります。筋生検をして、免疫組織学的染色でジスフェルリンが欠損していることを証明すれば診断は可能です。最近の研究ではどうも日本の肢帯型の患者さんの30%位はこのジスフェルリン欠損のようです。
最近注目を集めているのがLGMD2C−2Fで、いずれもジストロフィン結合蛋白であるサルコグリカン(サブユニットα、β、γ、δ)(図6参照)の遺伝子に変異があります。サルコグリカン欠損はLGMDの中でもかなり特徴ある臨床症状をとるので、サルコグリカノパチー(sarcoglycanopathy)と総称されています。デュシェンヌ型あるいはベッカー型によく似ていますが、常染色体劣性遺伝をとるので、男性と女性の比がほぼ同じです。
病理学的にはごく軽度の筋ジストロフィー変化から、強い変化をみるものまでと多彩です。デュシェンヌ型より慢性に経過するものが多いので、ベッカー型に似ています。肥大線維、筋線維の分割(fiber splitting)、中心核の増加をみます。また筋原線維間網の乱れ、高頻度に分葉線維(lobulated fiber)をみるのが特徴的で、この分葉線維はカルパイン3欠損に多いといわれています(図16参照)。
b.臨床症状
優性遺伝をとるものはきわめてまれで、日本ではまだ表のLGMD1の5型に相当する人はごく少数しか見つかっていませんので、ここでは常染色体劣性遺伝の患者さんについて述べます。
発症年齢は小児期から50歳代以降までと幅があります。病気の種類が多いのですから、症状もいろいろあるということはよく理解できます。筋力低下のような臨床症状はなく、検査上血清クレアチンキナーゼ(CK)値が高いだけという人もいます。一方たまにはデュシェンヌ型と変わらぬ重症経過をとるものもいますが、全体からみると病気はデュシェンヌ型より軽く、進行も遅いです。
近位筋(腰帯筋)が好んで侵されます。顔面筋罹患はありません。最初に気付かれる症状は歩行異常に関するものです。走れない、転びやすい、階段昇降困難などです。立ち上がるときに努力がいり、しばしばGowers徴候(立ち上がるときに膝に手をあてて立つ)をみます。下腿の仮性肥大はないか、あっても軽度です。
関節拘縮は下肢にみられ、早期から尖足をみるものもあります。歩行不能となると、全身の関節が拘縮するようになるのはデュシェンヌ型と同じです。ただし、心合併は少なく、呼吸不全も少ないので、生命的予後はよいとされています。
【附1】サルコグリカノパチー(sarcoglycanopathy)
ふくらはぎに仮性肥大があって、デュシェンヌ型に多少似るが、常染色体劣性遺伝をとる疾患が知られていました。それは本邦では悪性肢帯型筋ジストロフィー(三好)、欧米の教科書ではchildhood muscular dystrophy (Walton)などとよばれていたのです。Tunisia地方に多くみられる患者はsevere childhood autosomal recessive muscular dystrophy: SCARMDとして報告されています。これらの疾患はジストロフィン結合蛋白であるサルコグリカン複合体(図6参照)の欠損であることが明らかにされました。最も多いのはαサルコグリカン(adhalinともよばれる)欠損です。α、β、γ、δいずれのサブユニット遺伝子の変異も見つかっています。いずれのサブユニットの遺伝子の欠損でも、常染色体劣性遺伝をとり、臨床症状はよく似ています。臨床症状や筋生検からはサルコグリカノパチーの診断は容易です(図14)。しかし、どのサブユニット遺伝子変異であっても、αーδ全てのサブユニットの欠損をみるので、どのサブユニットの遺伝子の変異かはそれぞれの遺伝子を直接検査しないとわかりません。
【附2】カルパイン3異常症(カルパイノパチー)
カルパイン3とは筋構造蛋白の一つであるコネクチン(titinともよばれる)の調節酵素です。分子量94kdで、4つの主な働きをもつドメインをもっています。最近の国立精神・神経センターの研究では日本人の肢帯型筋ジストロフィーの約30%は本症であるとのデータがだされています。症状は比較的軽度で成人発症例もまれではありません。子どもでは血清CK値が高いだけで無症状の人もいます。臨床的にはこれといった診断的所見はありません(図15)。筋病理では比較的高率に分葉線維(lobulated fiber)をみます(図16)。
| 病型 | 遺伝子座 | 遺伝子産物 | |
| 常染色体優性遺伝 | LGMD 1A 1B 1C 1D 1E 1F | 5q31 1q11-21 3p25 6q23 7q 5q31 | myotilin lamin A/C caveloin3 ? ? ? |
| 常染色体劣性遺伝 | LGMD 2A 2B 2C 2D 2E 2F 2G 2H 2I | 15q15.1-q21.1 2p13 13q12 17q21 4q12 5q33-q34 17q11-q12 9q31-q34.1 19q13.3 | calpain 3 dysferlin γ-sarcoglycan α-sarcoglycan β-sarcoglycan δ-sarcoglycan telethonin ? FKRP |
| LGMD:limb-girdle muscular dystrophy | |||
| 表2:肢帯型筋ジストロフィーの分類 | |||
これからも次々と肢帯型の患者さんの中から新しい遺伝子の異常がみつかり、肢帯型筋ジストロフィーの種類はもっと増えるでしょう。表では優性遺伝は(5%以下とすくないのですが)LGMD1、劣性遺伝をとるものはLGMD2に分類されています。肢帯型といっても、その原因は多岐にわたることが明らかにされたのです。遺伝子診断や筋生検の免疫組織化学的染色により、肢帯型の患者さんの70-80%は近い将来、あの複雑な分類のどのタイプに当てはまるか決定できるようになると言われています。
a.病因、病態、病理
上で述べたように肢帯型は原因が多岐にわたっているので、筋生検で免疫組織学的染色をしたり、遺伝子解析をしても半数以上の人は原因が何か分かりません。優性遺伝をとるものには5つの型が知られています。日本ではこの5つの型に相当する病気を持った人は少数しか報告されていません。 常染色体劣性遺伝をとるLGMD2Aは蛋白分解酵素であるカルパイン3遺伝子に変異がみられています。カルパイン3という酵素は筋肉が収縮するときに必要な細い糸のような構造物の一つであるコネクチン(タイチンともいいます)を調節しています。しかし、この病気ではコネクチンは正常に発現されています。この酵素が異常となると、なぜ筋線維が壊死なるのかそれは不明です。いろいろ調べてみると肢帯型の患者さんの30%位はこのカルパイン3の遺伝子変異をもっていることが明らかにされています。カルパイン3遺伝子に変異をもった肢帯型のことはカルパイノパチーともよばれます。 LGMD2Bはジスフェルリン(dysferlin)という遺伝子に変異があり、ジスフェルリンが欠損する病気です。このジスフェリルンは筋細胞膜にあります。筋生検をして、免疫組織学的染色でジスフェルリンが欠損していることを証明すれば診断は可能です。最近の研究ではどうも日本の肢帯型の患者さんの30%位はこのジスフェルリン欠損のようです。
最近注目を集めているのがLGMD2C−2Fで、いずれもジストロフィン結合蛋白であるサルコグリカン(サブユニットα、β、γ、δ)(図6参照)の遺伝子に変異があります。サルコグリカン欠損はLGMDの中でもかなり特徴ある臨床症状をとるので、サルコグリカノパチー(sarcoglycanopathy)と総称されています。デュシェンヌ型あるいはベッカー型によく似ていますが、常染色体劣性遺伝をとるので、男性と女性の比がほぼ同じです。
病理学的にはごく軽度の筋ジストロフィー変化から、強い変化をみるものまでと多彩です。デュシェンヌ型より慢性に経過するものが多いので、ベッカー型に似ています。肥大線維、筋線維の分割(fiber splitting)、中心核の増加をみます。また筋原線維間網の乱れ、高頻度に分葉線維(lobulated fiber)をみるのが特徴的で、この分葉線維はカルパイン3欠損に多いといわれています(図16参照)。
b.臨床症状
優性遺伝をとるものはきわめてまれで、日本ではまだ表のLGMD1の5型に相当する人はごく少数しか見つかっていませんので、ここでは常染色体劣性遺伝の患者さんについて述べます。
発症年齢は小児期から50歳代以降までと幅があります。病気の種類が多いのですから、症状もいろいろあるということはよく理解できます。筋力低下のような臨床症状はなく、検査上血清クレアチンキナーゼ(CK)値が高いだけという人もいます。一方たまにはデュシェンヌ型と変わらぬ重症経過をとるものもいますが、全体からみると病気はデュシェンヌ型より軽く、進行も遅いです。
近位筋(腰帯筋)が好んで侵されます。顔面筋罹患はありません。最初に気付かれる症状は歩行異常に関するものです。走れない、転びやすい、階段昇降困難などです。立ち上がるときに努力がいり、しばしばGowers徴候(立ち上がるときに膝に手をあてて立つ)をみます。下腿の仮性肥大はないか、あっても軽度です。
関節拘縮は下肢にみられ、早期から尖足をみるものもあります。歩行不能となると、全身の関節が拘縮するようになるのはデュシェンヌ型と同じです。ただし、心合併は少なく、呼吸不全も少ないので、生命的予後はよいとされています。
【附1】サルコグリカノパチー(sarcoglycanopathy)
ふくらはぎに仮性肥大があって、デュシェンヌ型に多少似るが、常染色体劣性遺伝をとる疾患が知られていました。それは本邦では悪性肢帯型筋ジストロフィー(三好)、欧米の教科書ではchildhood muscular dystrophy (Walton)などとよばれていたのです。Tunisia地方に多くみられる患者はsevere childhood autosomal recessive muscular dystrophy: SCARMDとして報告されています。これらの疾患はジストロフィン結合蛋白であるサルコグリカン複合体(図6参照)の欠損であることが明らかにされました。最も多いのはαサルコグリカン(adhalinともよばれる)欠損です。α、β、γ、δいずれのサブユニット遺伝子の変異も見つかっています。いずれのサブユニットの遺伝子の欠損でも、常染色体劣性遺伝をとり、臨床症状はよく似ています。臨床症状や筋生検からはサルコグリカノパチーの診断は容易です(図14)。しかし、どのサブユニット遺伝子変異であっても、αーδ全てのサブユニットの欠損をみるので、どのサブユニットの遺伝子の変異かはそれぞれの遺伝子を直接検査しないとわかりません。
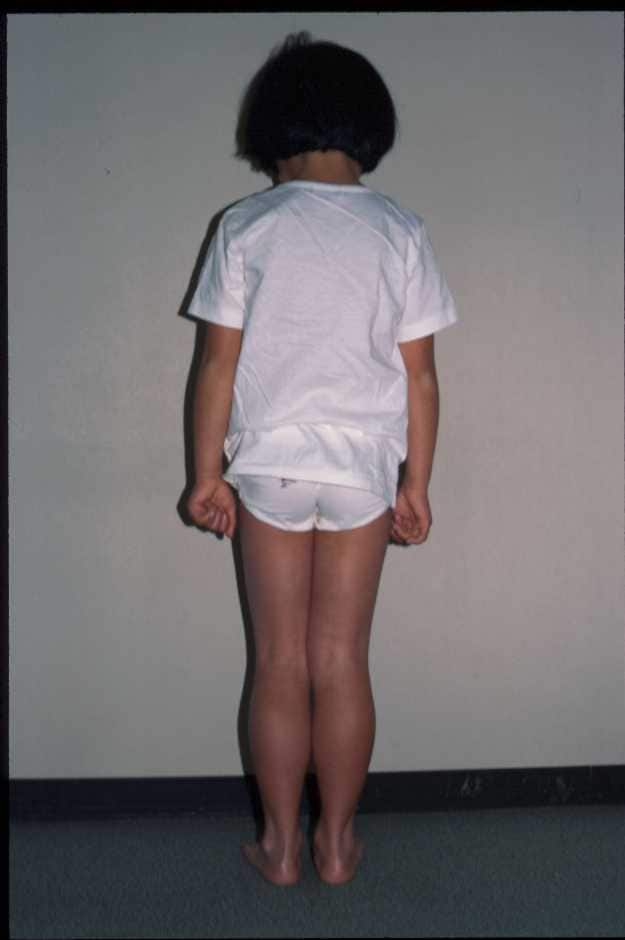 | 手足の力が4−5歳から弱くなり、ふくらはぎに偽性肥大をみ、デュシェンヌ型に似る。 筋生検でサルコグリカノパチーの診断をうけた。αサルコグリカン遺伝子に変異が認められた。 |
| 図14:サルコグリカノパチーの女児例 | |
カルパイン3とは筋構造蛋白の一つであるコネクチン(titinともよばれる)の調節酵素です。分子量94kdで、4つの主な働きをもつドメインをもっています。最近の国立精神・神経センターの研究では日本人の肢帯型筋ジストロフィーの約30%は本症であるとのデータがだされています。症状は比較的軽度で成人発症例もまれではありません。子どもでは血清CK値が高いだけで無症状の人もいます。臨床的にはこれといった診断的所見はありません(図15)。筋病理では比較的高率に分葉線維(lobulated fiber)をみます(図16)。
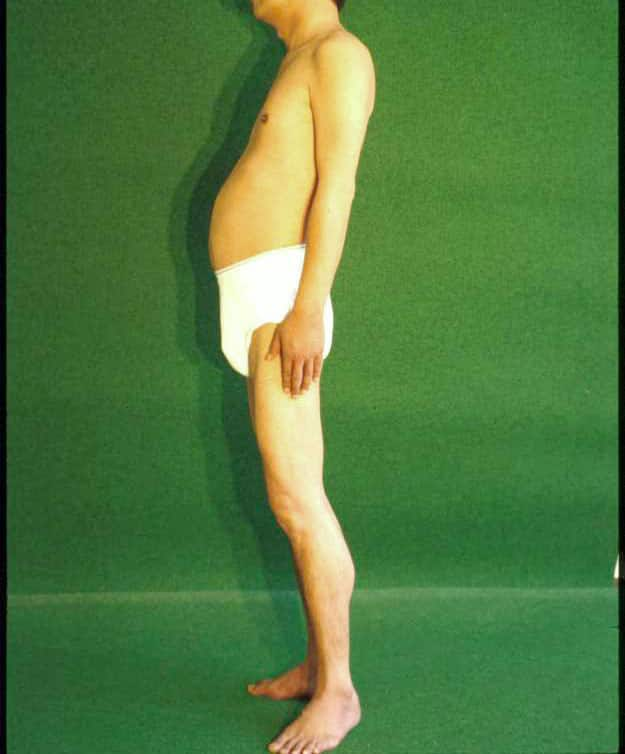 | 筋力低下は軽く、進行は遅い。 ときにふくらはぎに偽性肥大をみる。 この患者さんは躯幹筋が特に弱いので前彎がみられる。 |
| 図15:カルパイノパチーの成人例 | |
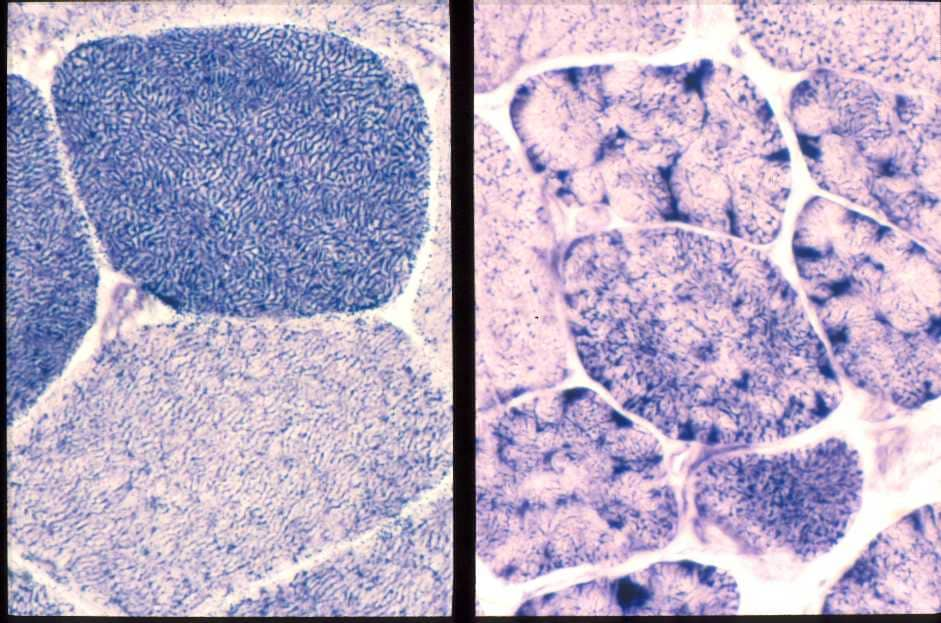 | この写真のようなNADH-TRという特殊な染色をすると、正常人(左)ではタイプ1線維は濃く染まり、筋線維の中の網目構造がよくわかる。カルパイノパチーの人(右)では筋線維は細く、クローバの葉の様にみえる分葉線維が増加している。 |
| 図16:カルパイノパチーの筋病理 |
- LGMDのページへのリンク
